

Reconstructing the regulatory programs underlying the phenotypic plasticity of neural cancers
- PMID: 39516198
- PMCID: PMC11549355
- DOI: 10.1038/s41467-024-53954-3
Reconstructing the regulatory programs underlying the phenotypic plasticity of neural cancers
Abstract
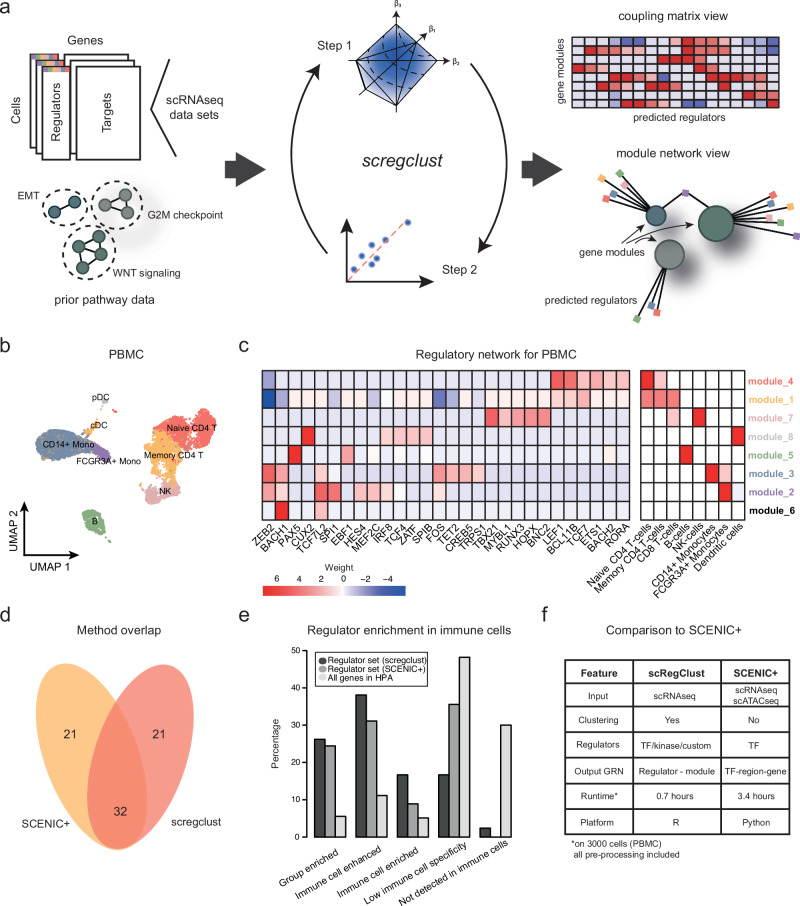
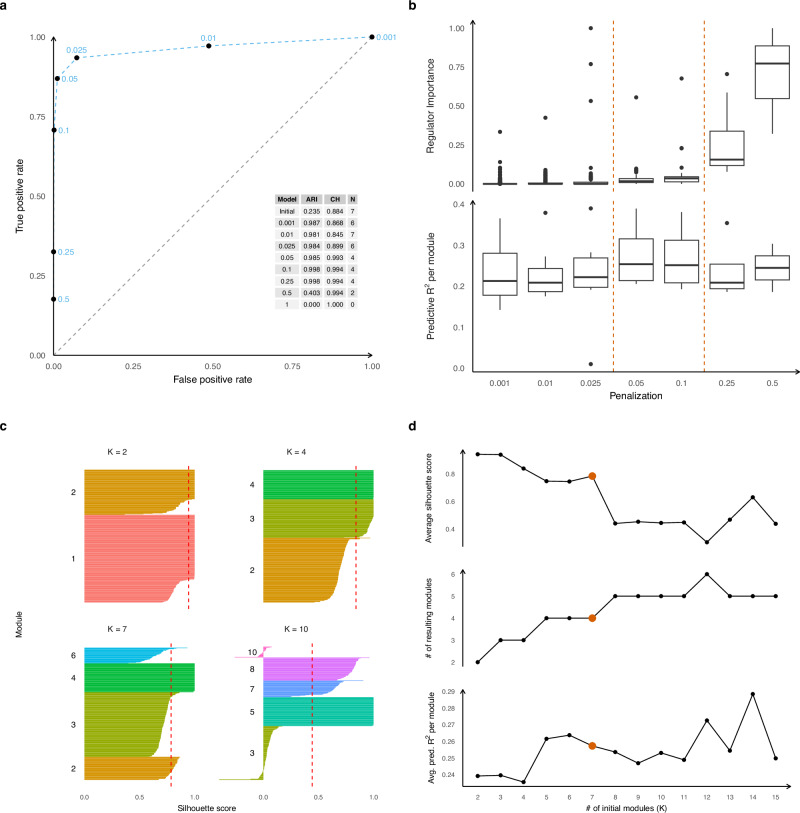
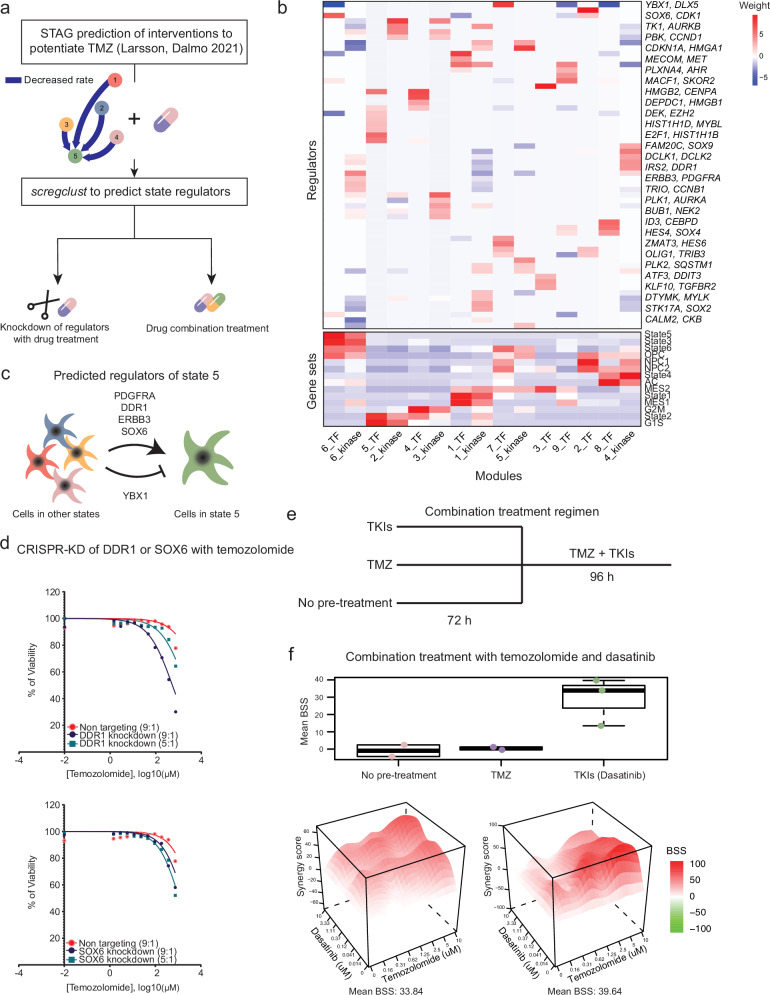
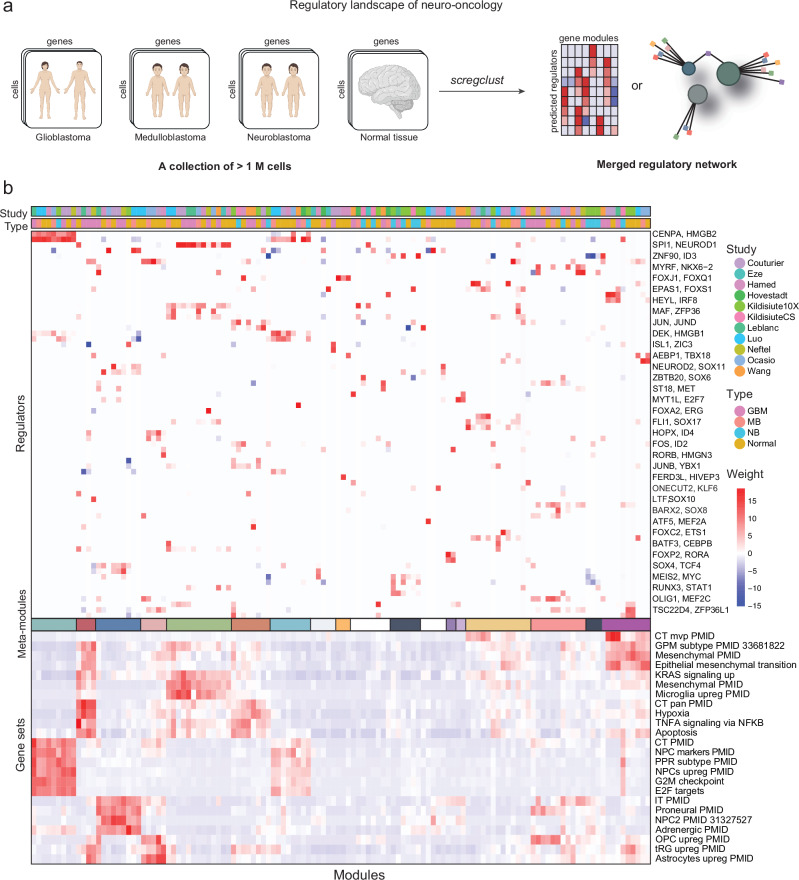
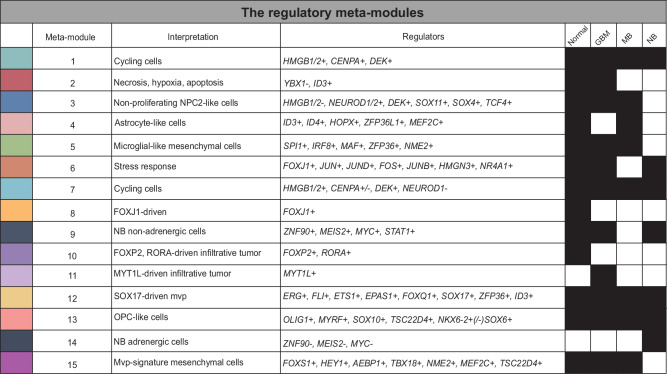
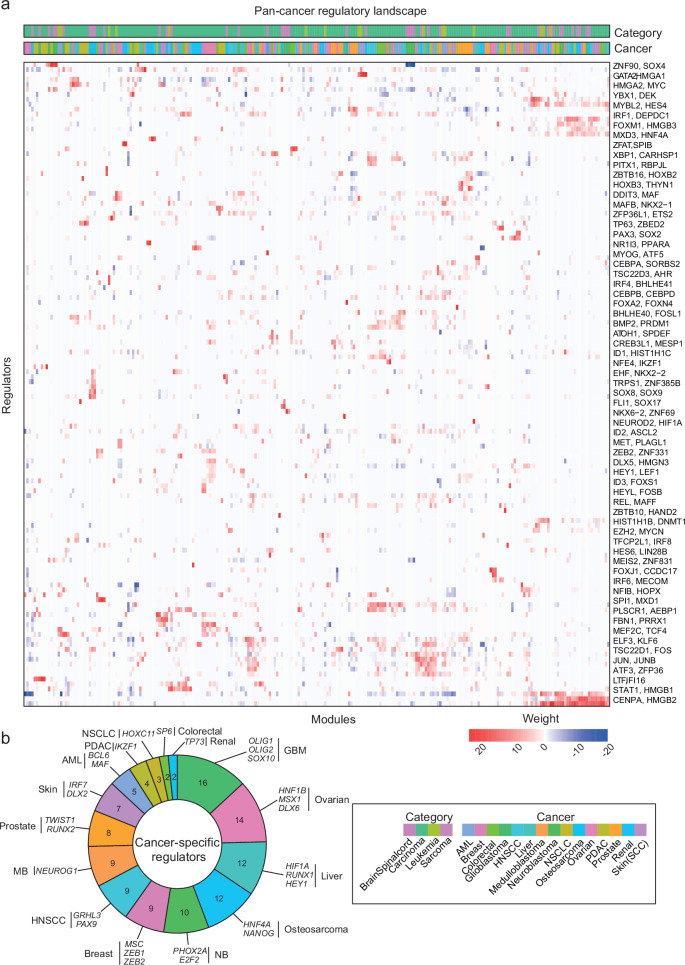
Nervous system cancers exhibit diverse transcriptional cell states influenced by normal development, injury response, and growth. However, the understanding of these states' regulation and pharmacological relevance remains limited. Here we present "single-cell regulatory-driven clustering" (scregclust), a method that reconstructs cellular regulatory programs from extensive collections of single-cell RNA sequencing (scRNA-seq) data from both tumors and developing tissues. The algorithm efficiently divides target genes into modules, predicting key transcription factors and kinases with minimal computational time. Applying this method to adult and childhood brain cancers, we identify critical regulators and suggest interventions that could improve temozolomide treatment in glioblastoma. Additionally, our integrative analysis reveals a meta-module regulated by SPI1 and IRF8 linked to an immune-mediated mesenchymal-like state. Finally, scregclust's flexibility is demonstrated across 15 tumor types, uncovering both pan-cancer and specific regulators. The algorithm is provided as an easy-to-use R package that facilitates the exploration of regulatory programs underlying cell plasticity.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Richards, L. M. et al. Gradient of developmental and injury response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer2, 157–173 (2021). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- SRA/PRJNA637987
- figshare/10.6084/m9.figshare.25909156
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
