

SARS-CoV-2 Displays a Suboptimal Codon Usage Bias for Efficient Translation in Human Cells Diverted by Hijacking the tRNA Epitranscriptome
- PMID: 39519170
- PMCID: PMC11546939
- DOI: 10.3390/ijms252111614
SARS-CoV-2 Displays a Suboptimal Codon Usage Bias for Efficient Translation in Human Cells Diverted by Hijacking the tRNA Epitranscriptome
Abstract
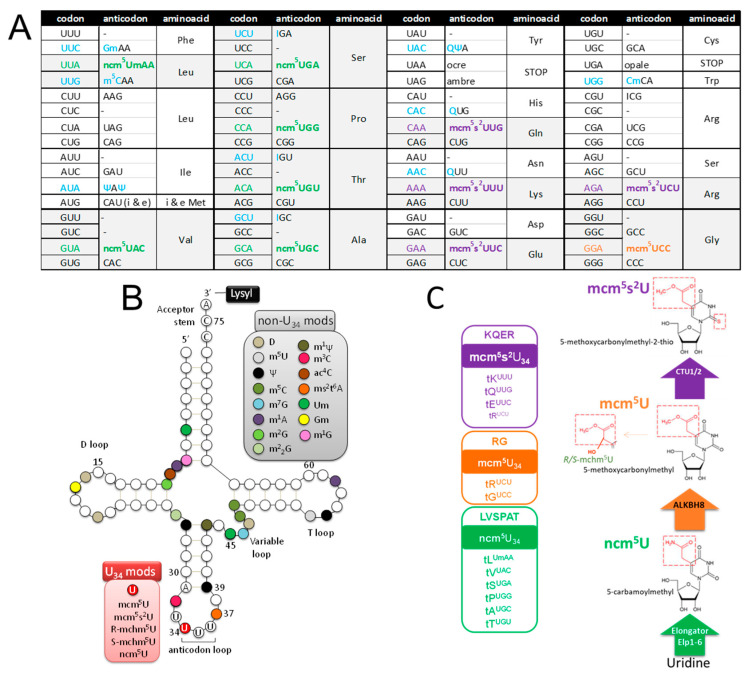
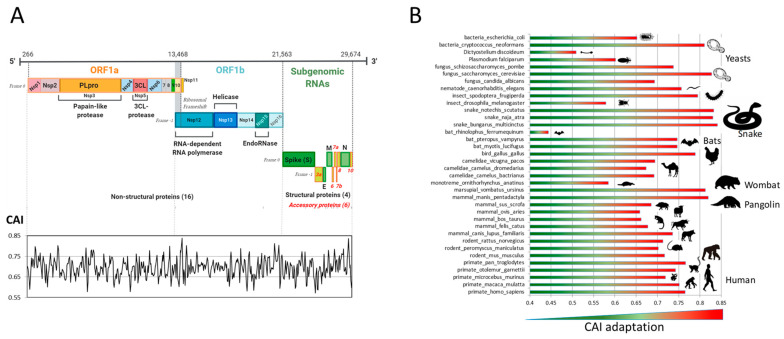
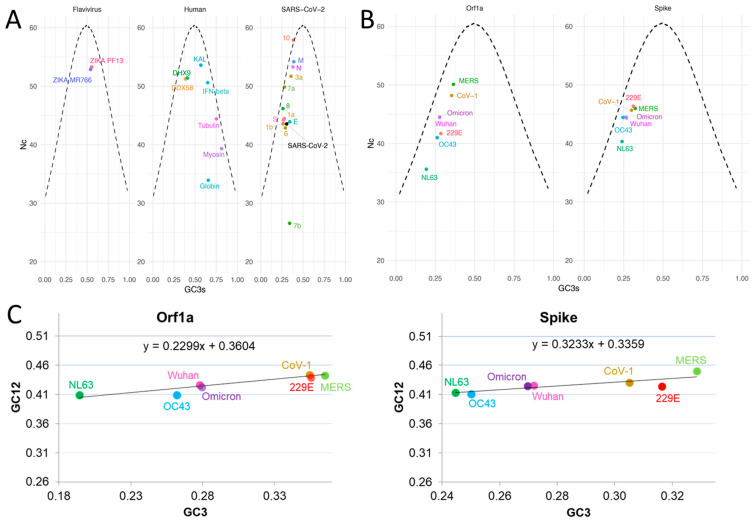
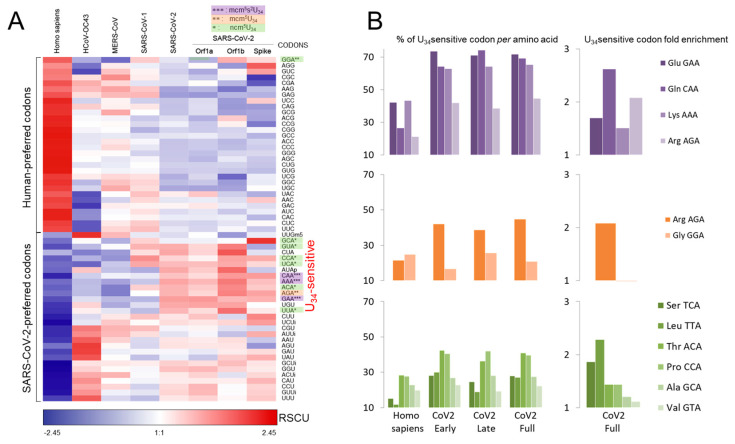
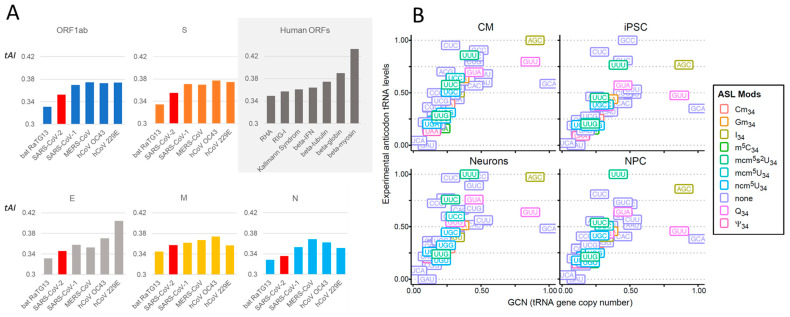
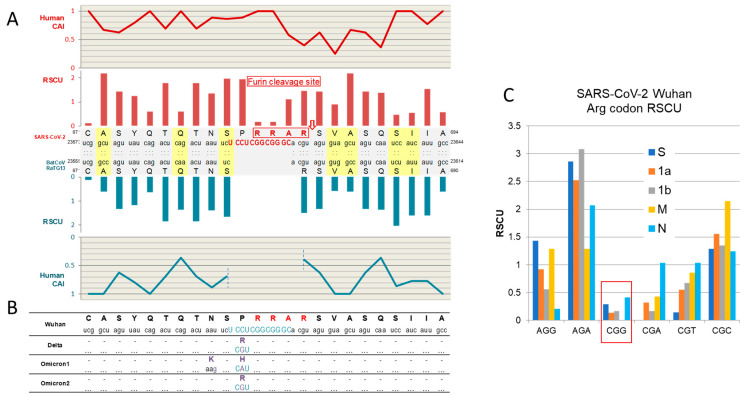
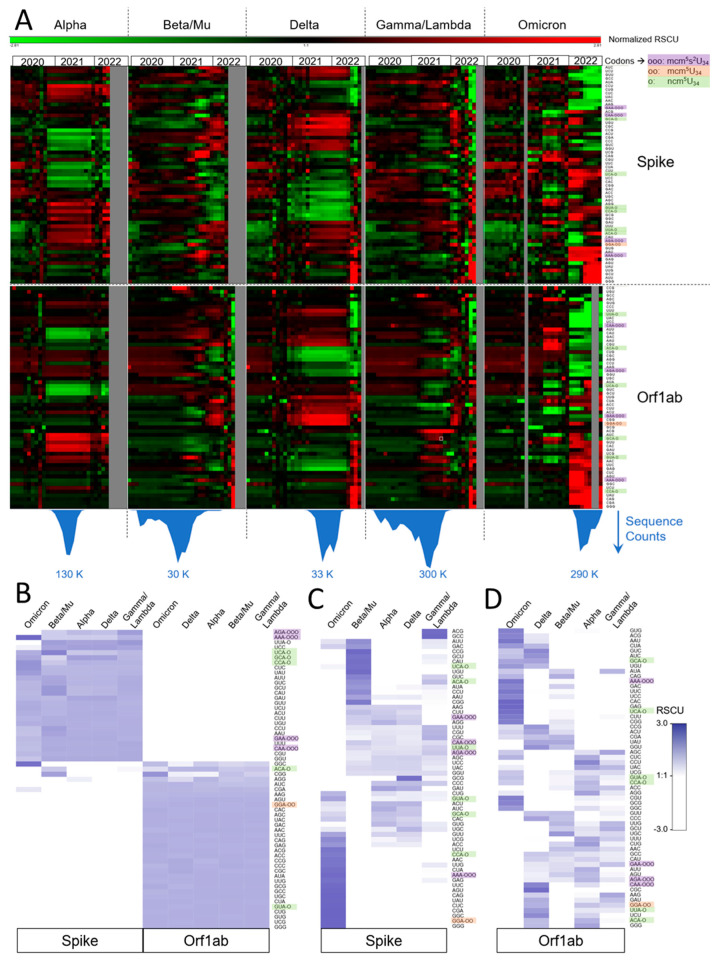
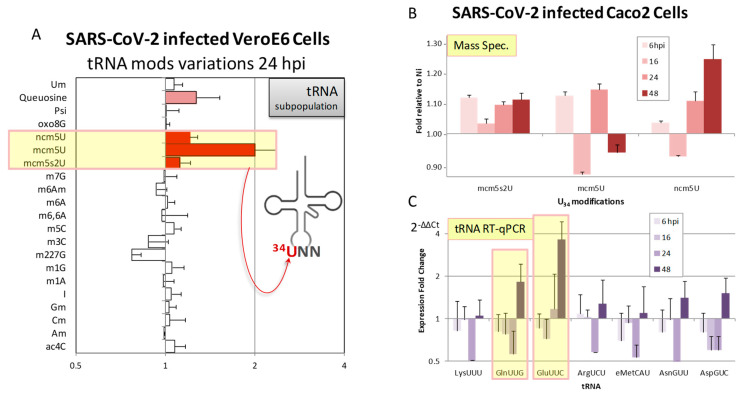
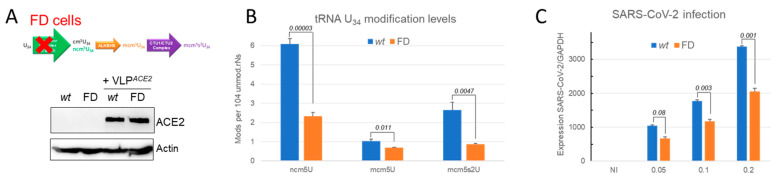
Codon bias analysis of SARS-CoV-2 reveals suboptimal adaptation for translation in human cells it infects. The detailed examination of the codons preferentially used by SARS-CoV-2 shows a strong preference for LysAAA, GlnCAA, GluGAA, and ArgAGA, which are infrequently used in human genes. In the absence of an adapted tRNA pool, efficient decoding of these codons requires a 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2) modification at the U34 wobble position of the corresponding tRNAs (tLysUUU; tGlnUUG; tGluUUC; tArgUCU). The optimal translation of SARS-CoV-2 open reading frames (ORFs) may therefore require several adjustments to the host's translation machinery, enabling the highly biased viral genome to achieve a more favorable "Ready-to-Translate" state in human cells. Experimental approaches based on LC-MS/MS quantification of tRNA modifications and on alteration of enzymatic tRNA modification pathways provide strong evidence to support the hypothesis that SARS-CoV-2 induces U34 tRNA modifications and relies on these modifications for its lifecycle. The conclusions emphasize the need for future studies on the evolution of SARS-CoV-2 codon bias and its ability to alter the host tRNA pool through the manipulation of RNA modifications.
Keywords: SARS-CoV-2; codon usage; epitranscriptome; tRNA; translation.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Molecular Coping Mechanisms: Reprogramming tRNAs To Regulate Codon-Biased Translation of Stress Response Proteins.Acc Chem Res. 2023 Dec 5;56(23):3504-3514. doi: 10.1021/acs.accounts.3c00572. Epub 2023 Nov 22. Acc Chem Res. 2023. PMID: 37992267 Free PMC article.
-
tRNA Modifications: A Tale of Two Viruses-SARS-CoV-2 and ZIKV.Int J Mol Sci. 2025 Aug 2;26(15):7479. doi: 10.3390/ijms26157479. Int J Mol Sci. 2025. PMID: 40806605 Free PMC article.
-
Betacoronaviruses genome analysis reveals evolution toward specific codons usage: Implications for SARS-CoV-2 mitigation strategies.J Med Virol. 2021 Sep;93(9):5630-5634. doi: 10.1002/jmv.27056. Epub 2021 May 24. J Med Virol. 2021. PMID: 33934387 Free PMC article.
-
Dysfunctional tRNA reprogramming and codon-biased translation in cancer.Trends Mol Med. 2022 Nov;28(11):964-978. doi: 10.1016/j.molmed.2022.09.007. Epub 2022 Oct 11. Trends Mol Med. 2022. PMID: 36241532 Free PMC article. Review.
-
Sulfur Modifications of the Wobble U34 in tRNAs and their Intracellular Localization in Eukaryotic Cells.Biomolecules. 2017 Feb 18;7(1):17. doi: 10.3390/biom7010017. Biomolecules. 2017. PMID: 28218716 Free PMC article. Review.
Cited by
-
ORF1ab codon frequency model predicts host-pathogen relationship in orthocoronavirinae.Front Bioinform. 2025 Mar 18;5:1562668. doi: 10.3389/fbinf.2025.1562668. eCollection 2025. Front Bioinform. 2025. PMID: 40170904 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
