

Aurora B inhibitors promote RB hypophosphorylation and senescence independent of p53-dependent CDK2/4 inhibition
- PMID: 39521795
- PMCID: PMC11550316
- DOI: 10.1038/s41419-024-07204-5
Aurora B inhibitors promote RB hypophosphorylation and senescence independent of p53-dependent CDK2/4 inhibition
Abstract
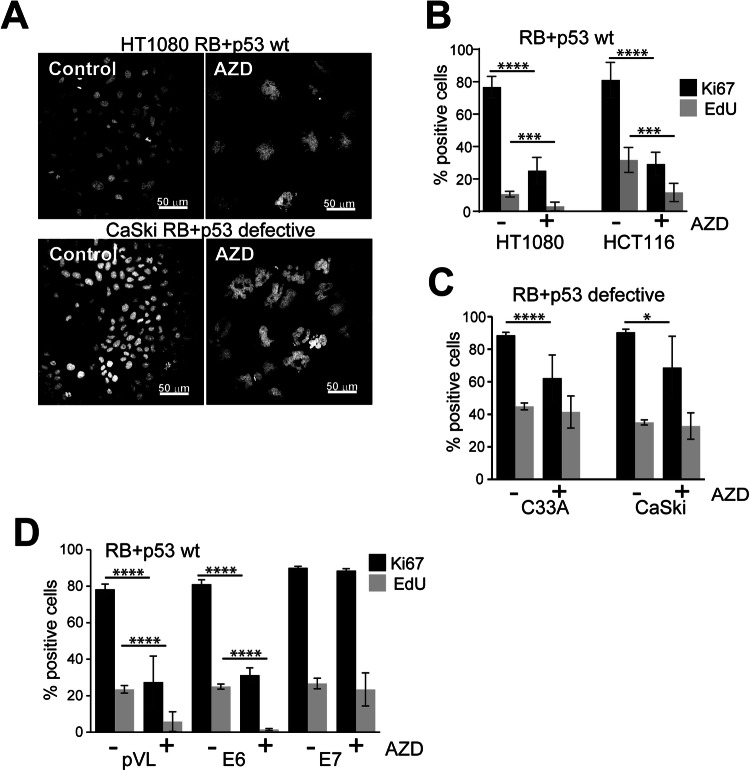
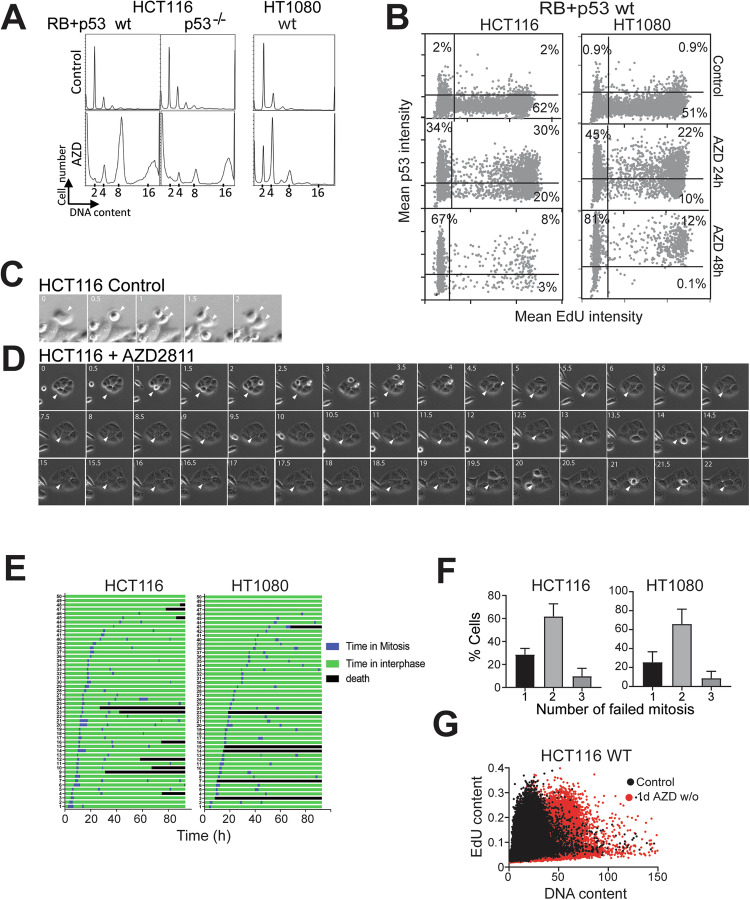
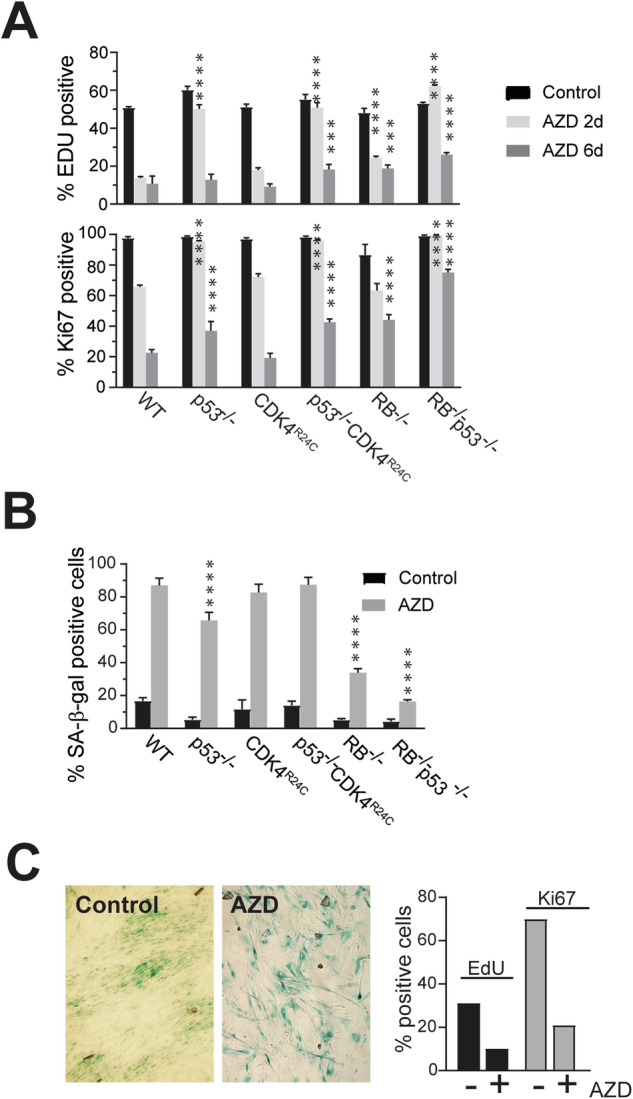
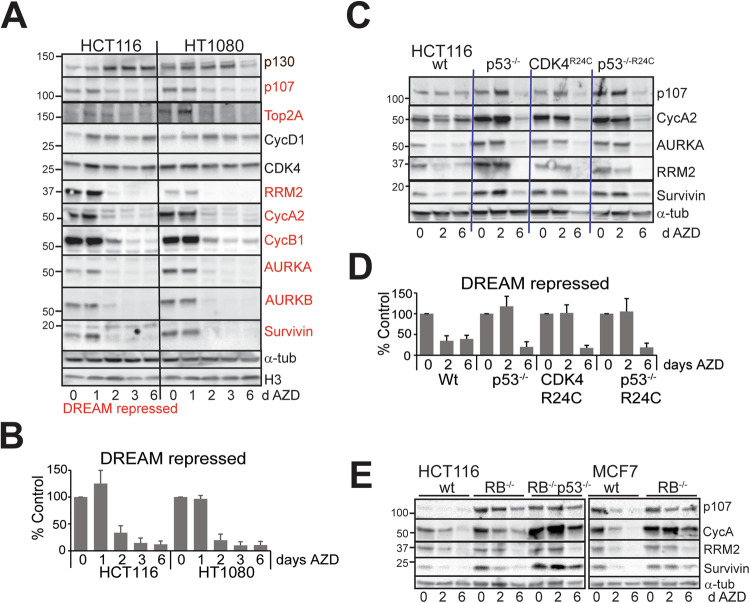
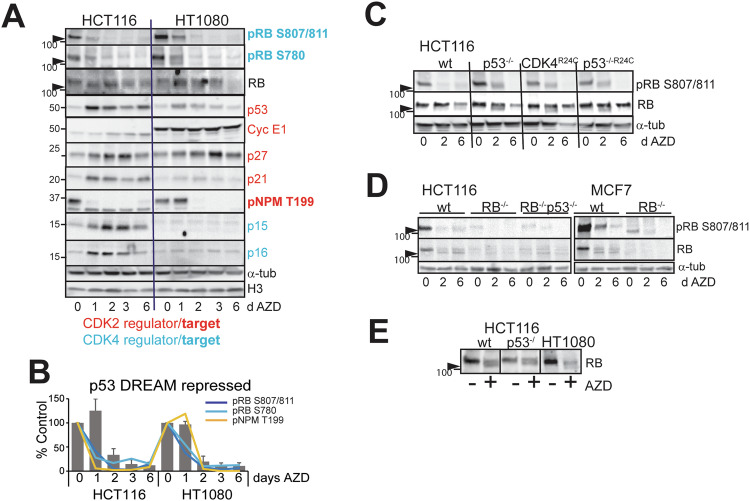
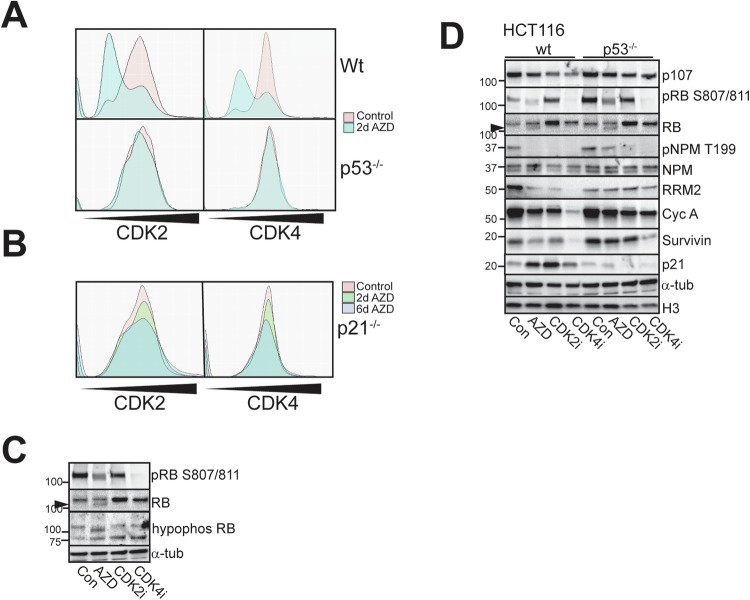
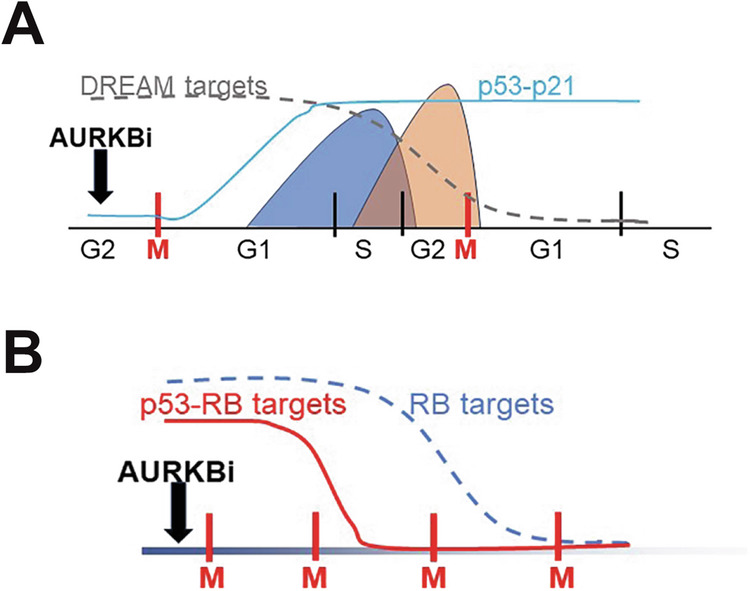
Aurora B kinase (AURKB) inhibitors have been trialled in a range of different tumour types but are not approved for any indication. Expression of the human papilloma virus (HPV) oncogenes and loss of retinoblastoma (RB) protein function has been reported to increase sensitivity to AURKB inhibitors but the mechanism of their contribution to sensitivity is poorly understood. Two commonly reported outcomes of AURKB inhibition are polyploidy and senescence, although their relationship is unclear. Here we have investigated the major cellular targets of the HPV E6 and E7, p53 and RB, to determine their contribution to AURKB inhibitor induced polyploidy and senescence. We demonstrate that polyploidy is a universal feature of AURKB inhibitor treatment in all cell types including normal primary cells, but the subsequent outcomes are controlled by RB and p53. We demonstrate that p53 by regulating p21 expression is required for an initial cell cycle arrest by inhibiting both CDK2 and CDK4 activity, but this arrest is only triggered after cells have undergone two failed mitosis and cytokinesis. However, cells can enter senescence in the absence of p53. RB is essential for AURKB inhibitor-induced senescence. AURKB inhibitor induces rapid hypophosphorylation of RB independent of inhibition of CDK2 or CDK4 kinases and p53. This work demonstrates that p53 activation determines the timing of senescence onset, but RB is indispensable for senescence.
© 2024. The Author(s).
Conflict of interest statement
JT, JS and JU are current or former employees and shareholders of AstraZeneca. Other authors declare no conflict of interest
Figures
References
-
- Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60–72. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
