

Multi-modal analysis of human hepatic stellate cells identifies novel therapeutic targets for metabolic dysfunction-associated steatotic liver disease
- PMID: 39522884
- PMCID: PMC12306656
- DOI: 10.1016/j.jhep.2024.10.044
Multi-modal analysis of human hepatic stellate cells identifies novel therapeutic targets for metabolic dysfunction-associated steatotic liver disease
Abstract
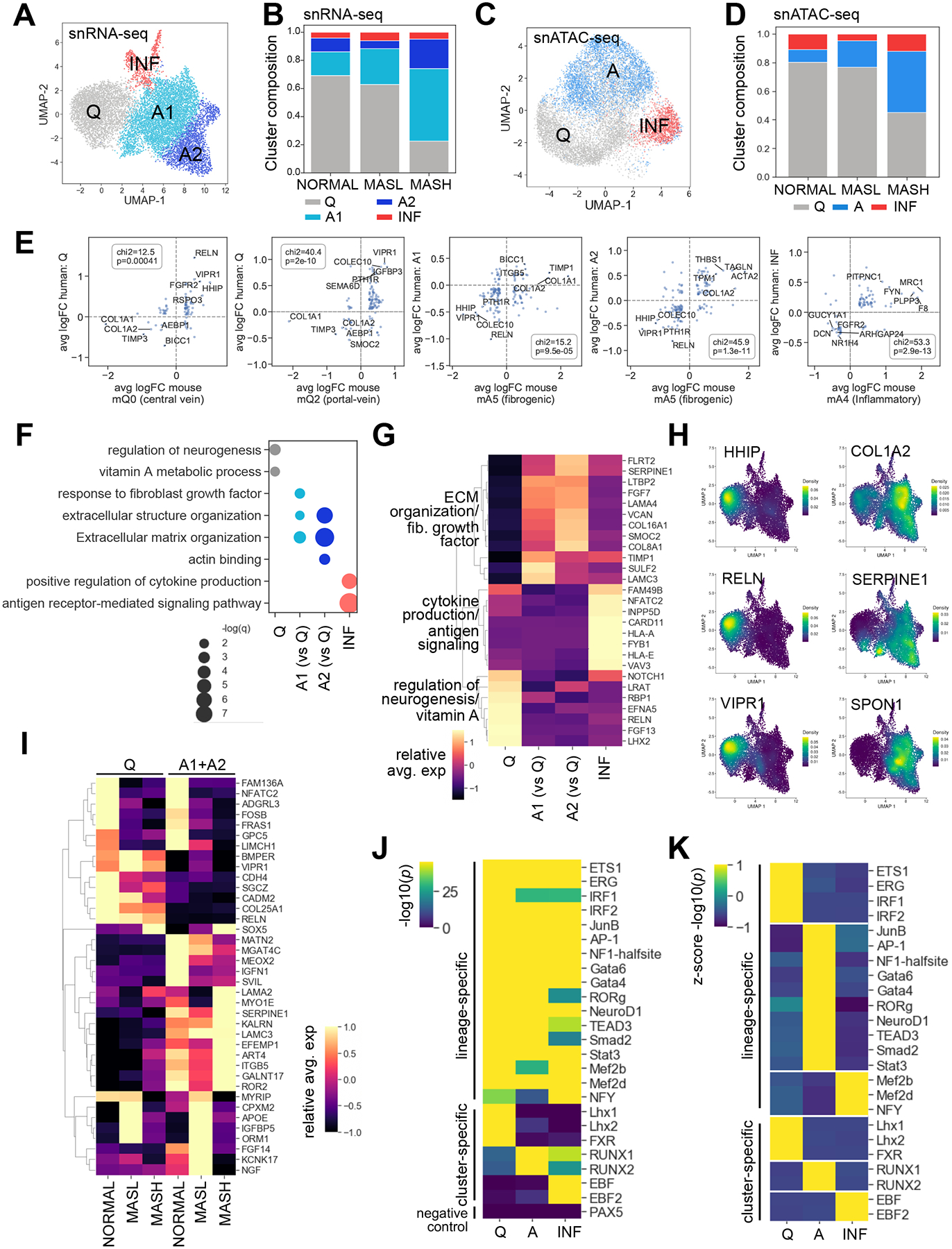
Background & aims: Metabolic dysfunction-associated steatotic liver disease ranges from metabolic dysfunction-associated steatotic liver (MASL) to metabolic dysfunction-associated steatohepatitis (MASH) with fibrosis. Transdifferentiation of hepatic stellate cells (HSCs) into fibrogenic myofibroblasts plays a critical role in the pathogenesis of MASH liver fibrosis. We compared transcriptome and chromatin accessibility of human HSCs from NORMAL, MASL, and MASH livers at single-cell resolution. We aimed to identify genes that are upregulated in activated HSCs and to determine which of these genes are key in the pathogenesis of MASH fibrosis.
Methods: Eighteen human livers were profiled using single-nucleus (sn)RNA-seq and snATAC-seq. High priority targets were identified, then tested in 2D human HSC cultures, 3D human liver spheroids, and HSC-specific gene knockout mice.
Results: MASH-enriched activated HSC subclusters are the major source of extracellular matrix proteins. We identified a set of concurrently upregulated and more accessible core genes (GAS7, SPON1, SERPINE1, LTBP2, KLF9, EFEMP1) that drive activation of HSCs. Expression of these genes was regulated via crosstalk between lineage-specific (JUNB/AP1), cluster-specific (RUNX1/2) and signal-specific (FOXA1/2) transcription factors. The pathological relevance of the selected targets, such as SERPINE1 (PAI-1), was demonstrated using dsiRNA-based HSC-specific gene knockdown or pharmacological inhibition of PAI-1 in 3D human MASH liver spheroids, and HSC-specific Serpine1 knockout mice.
Conclusion: This study identified novel gene targets and regulatory mechanisms underlying activation of fibrogenic HSCs in MASH, and demonstrated that genetic or pharmacological inhibition of select genes suppressed liver fibrosis.
Impact and implications: Herein, we present the results of a multi-modal sequencing analysis of human hepatic stellate cells (HSCs) from NORMAL, MASL (metabolic dysfunction-associated steatotic liver), and metabolic dysfunction-associated steatohepatitis (MASH) livers. We identified additional subclusters that were not detected by previous studies and characterized the mechanism by which HSCs are activated in MASH livers, including the transcriptional machinery that induces the transdifferentiation of HSCs into myofibroblasts. For the first time, we described the pathogenic role of activated HSC-derived PAI-1 (a product of the SERPINE1 gene) in the development of MASH liver fibrosis. Targeting the RUNX1/2-SERPINE1 axis could be a novel strategy for the treatment of liver fibrosis in patients.
Keywords: MASH; MASLD; activation of human HSCs; liver fibrosis; snRNA-seq and snATAC-seq.
Copyright © 2024 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Conflict of interest The authors of this study declare that they do not have any conflict of interest. Please refer to the accompanying ICMJE disclosure forms for further details.
Figures






References
-
- Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nature Reviews Gastroenterology & Hepatology 2021;18:151–166. - PubMed
MeSH terms
Grants and funding
- UL1 TR001442/TR/NCATS NIH HHS/United States
- P42 ES010337/ES/NIEHS NIH HHS/United States
- P30 DK120515/DK/NIDDK NIH HHS/United States
- U01 AA029019/AA/NIAAA NIH HHS/United States
- R01 AA028550/AA/NIAAA NIH HHS/United States
- U01 AA022614/AA/NIAAA NIH HHS/United States
- P50 AA011999/AA/NIAAA NIH HHS/United States
- R56 DK088837/DK/NIDDK NIH HHS/United States
- R01 DK091183/DK/NIDDK NIH HHS/United States
- R01 CA285997/CA/NCI NIH HHS/United States
- R01 DK111866/DK/NIDDK NIH HHS/United States
- R01 DK099205/DK/NIDDK NIH HHS/United States
- R01 DK101737/DK/NIDDK NIH HHS/United States
- R44 DK115242/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
