

The pro-oncogenic noncanonical activity of a RAS•GTP:RanGAP1 complex facilitates nuclear protein export
- PMID: 39528835
- PMCID: PMC11663792
- DOI: 10.1038/s43018-024-00847-5
The pro-oncogenic noncanonical activity of a RAS•GTP:RanGAP1 complex facilitates nuclear protein export
Abstract
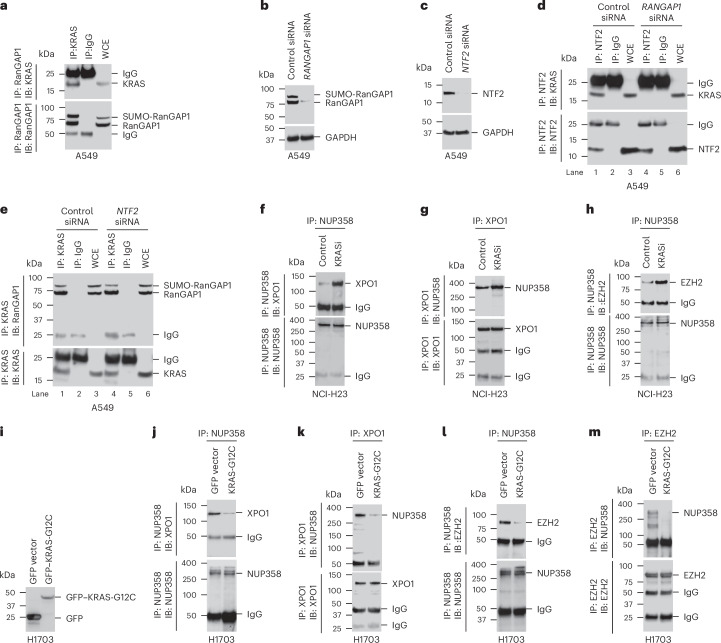
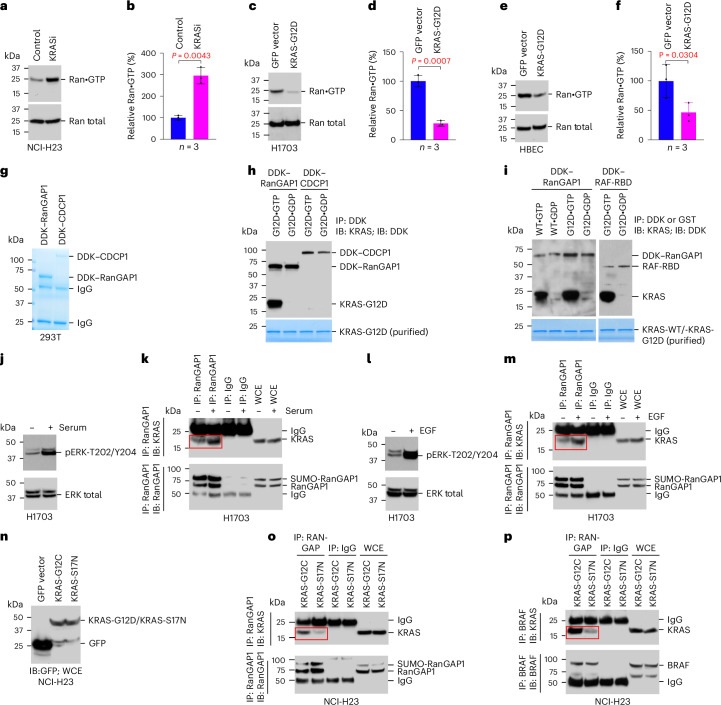
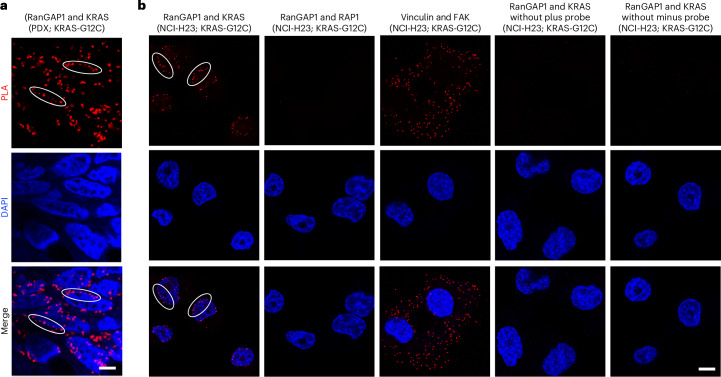
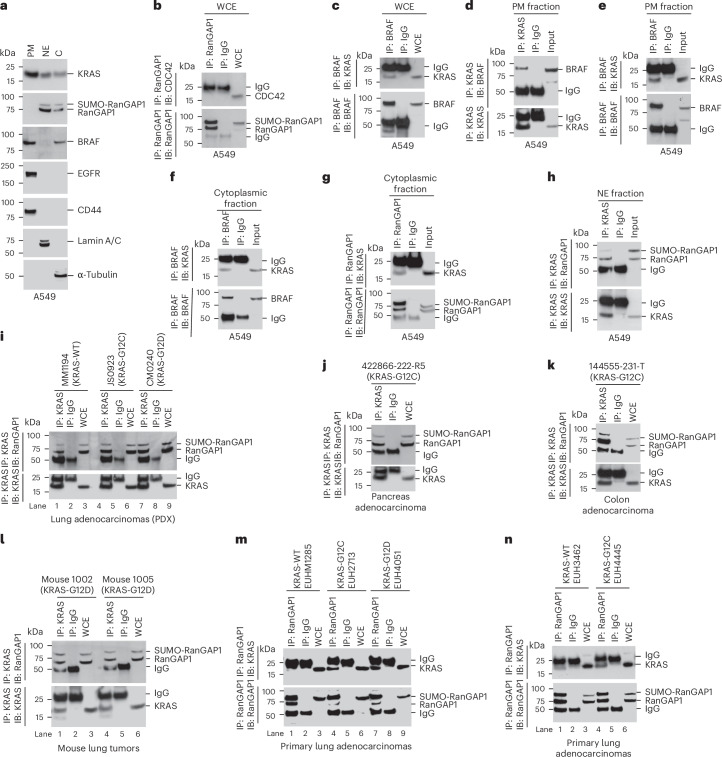
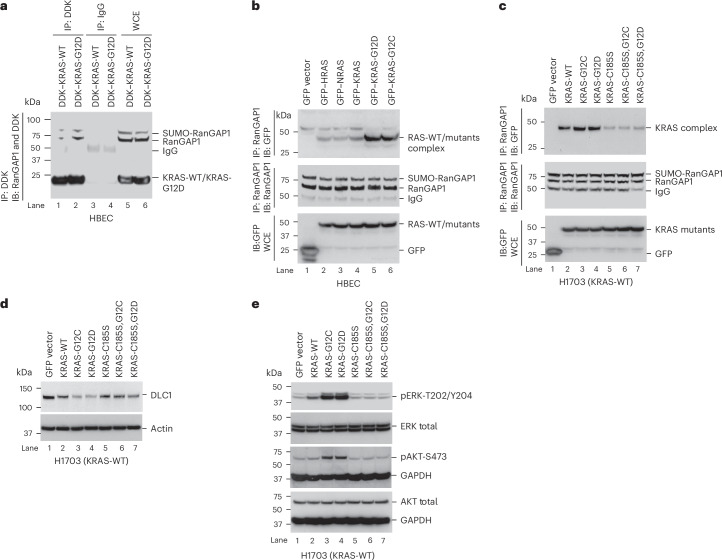
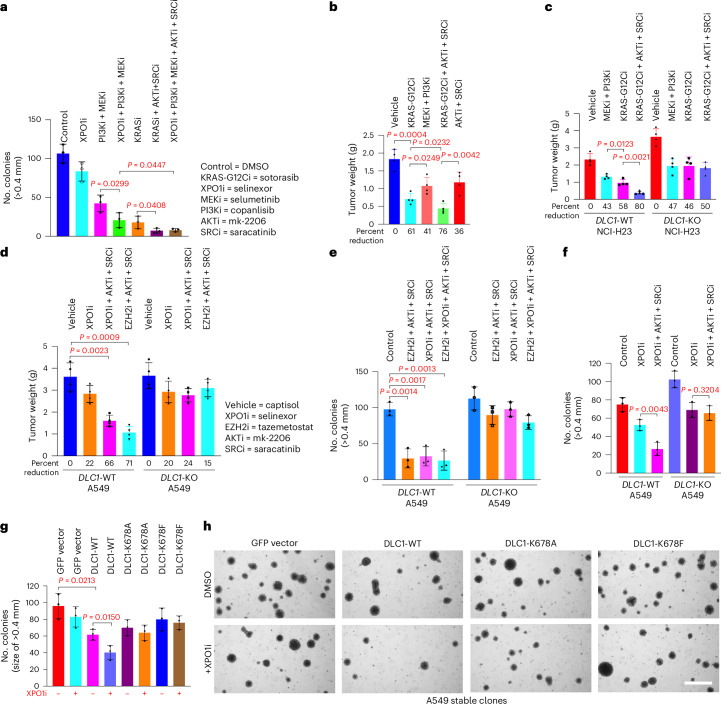
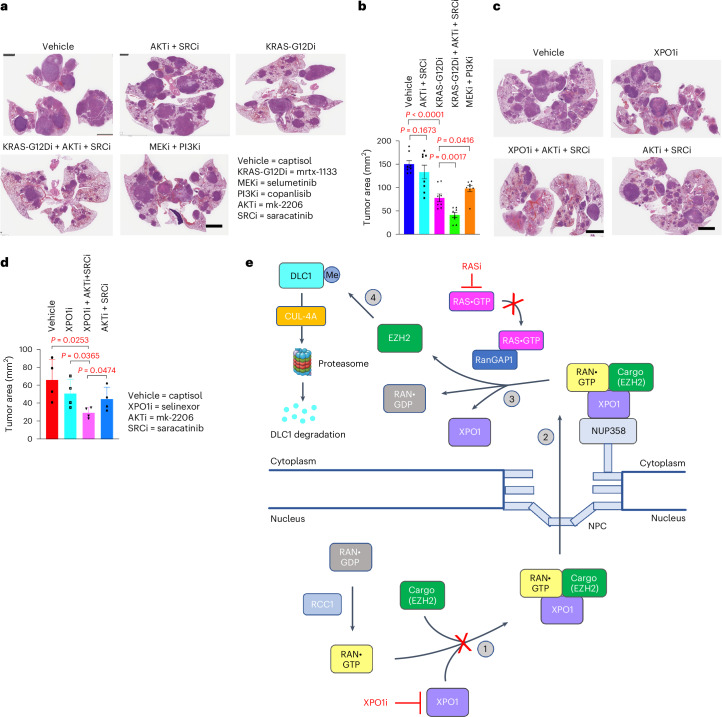
Canonical RAS signaling, including PI3K/AKT- and RAF/MEK-dependent activities, results mainly from RAS•GTP interaction with its effectors at the plasma membrane. Here, we identified a fundamental, oncogenic, noncanonical RAS•GTP activity that increases XPO1-dependent export of nuclear protein cargo into the cytoplasm and is independent of PI3K/AKT and RAF/MEK signaling. This RAS-dependent step acts downstream from XPO1 binding to nuclear protein cargo and is mediated by a perinuclear protein complex between RAS•GTP and RanGAP1 that facilitates hydrolysis of Ran•GTP to Ran•GDP, which promotes release of nuclear protein cargo into the cytoplasm. The export of nuclear EZH2, which promotes cytoplasmic degradation of the DLC1 tumor suppressor protein, is a biologically important component of this pro-oncogenic activity. Conversely, preventing nuclear protein export contributes to the antitumor activity of KRAS inhibition, which can be further augmented by reactivating the tumor suppressor activity of DLC1 or potentially combining RAS inhibitors with other cancer treatments.
© 2024. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Competing interests: All authors declare no competing interests.
Figures









References
-
- Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov.12, 31–46 (2022). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
