Laboratory validation of a clinical metagenomic next-generation sequencing assay for respiratory virus detection and discovery
- PMID: 39532844
- PMCID: PMC11558004
- DOI: 10.1038/s41467-024-51470-y
Laboratory validation of a clinical metagenomic next-generation sequencing assay for respiratory virus detection and discovery
Abstract
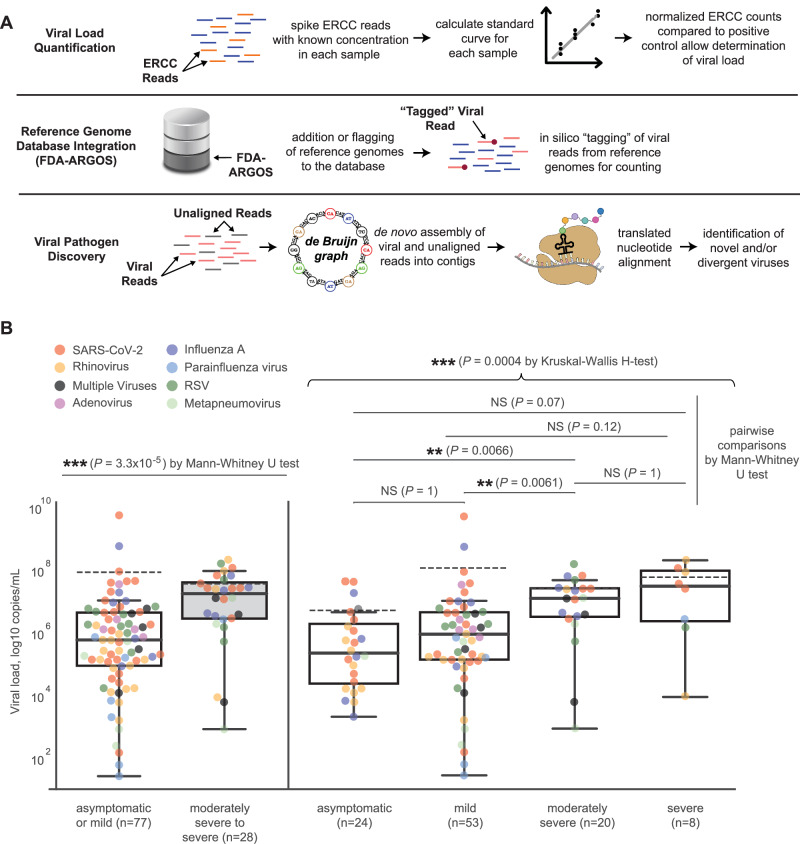
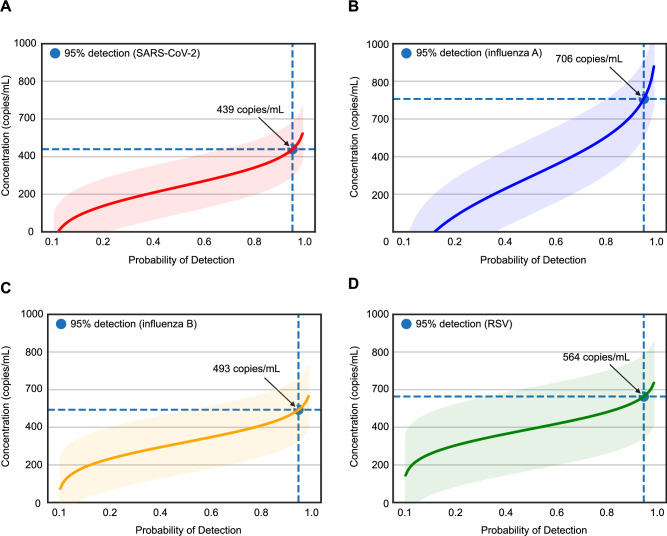
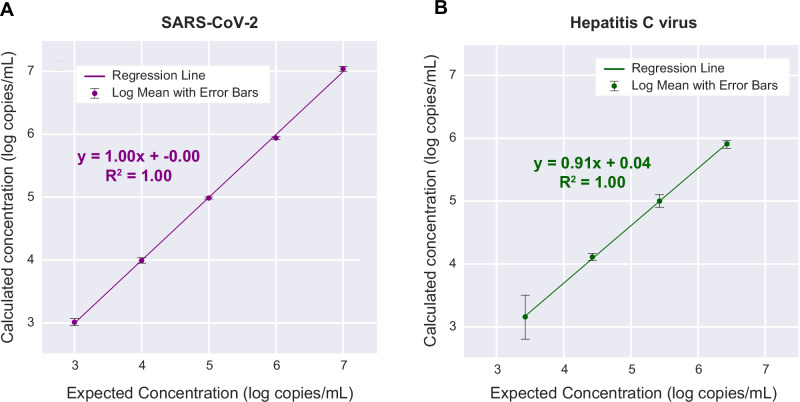
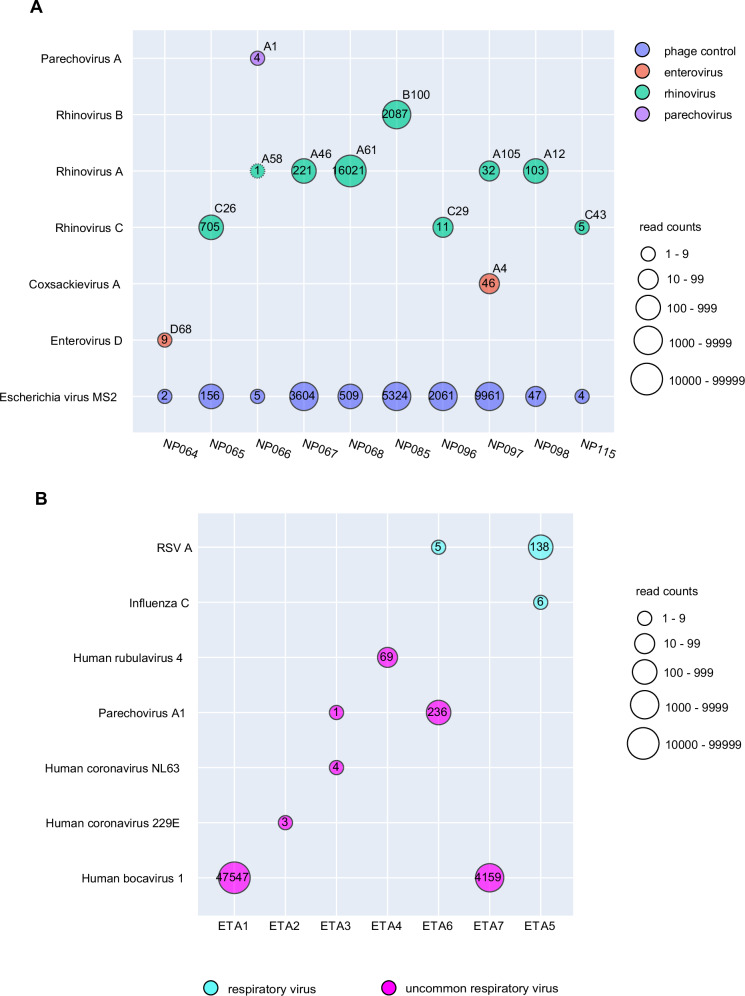
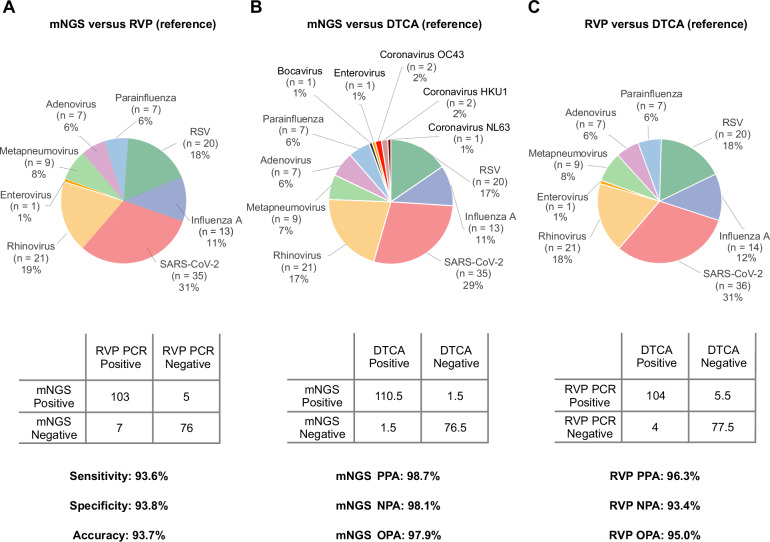
Tools for rapid identification of novel and/or emerging viruses are urgently needed for clinical diagnosis of unexplained infections and pandemic preparedness. Here we developed and clinically validated a largely automated metagenomic next-generation sequencing (mNGS) assay for agnostic detection of respiratory viral pathogens from upper respiratory swab and bronchoalveolar lavage samples in <24 h. The mNGS assay achieved mean limits of detection of 543 copies/mL, viral load quantification with 100% linearity, and 93.6% sensitivity, 93.8% specificity, and 93.7% accuracy compared to gold-standard clinical multiplex RT-PCR testing. Performance increased to 97.9% overall predictive agreement after discrepancy testing and clinical adjudication, which was superior to that of RT-PCR (95.0% agreement). To enable discovery of novel, sequence-divergent human viruses with pandemic potential, de novo assembly and translated nucleotide algorithms were incorporated into the automated SURPI+ computational pipeline used by the mNGS assay for pathogen detection. Using in silico analysis, we showed that after removal of all human viral sequences from the reference database, 70 (100%) of 70 representative human viral pathogens could still be identified based on homology to related animal or plant viruses. Our assay, which was granted breakthrough device designation from the US Food and Drug Administration (FDA) in August of 2023, demonstrates the feasibility of routine mNGS testing in clinical and public health laboratories, thus facilitating a robust and rapid response to the next viral pandemic.
© 2024. The Author(s).
Conflict of interest statement
Figures