

Structural basis of MICAL autoinhibition
- PMID: 39532862
- PMCID: PMC11557892
- DOI: 10.1038/s41467-024-54131-2
Structural basis of MICAL autoinhibition
Abstract
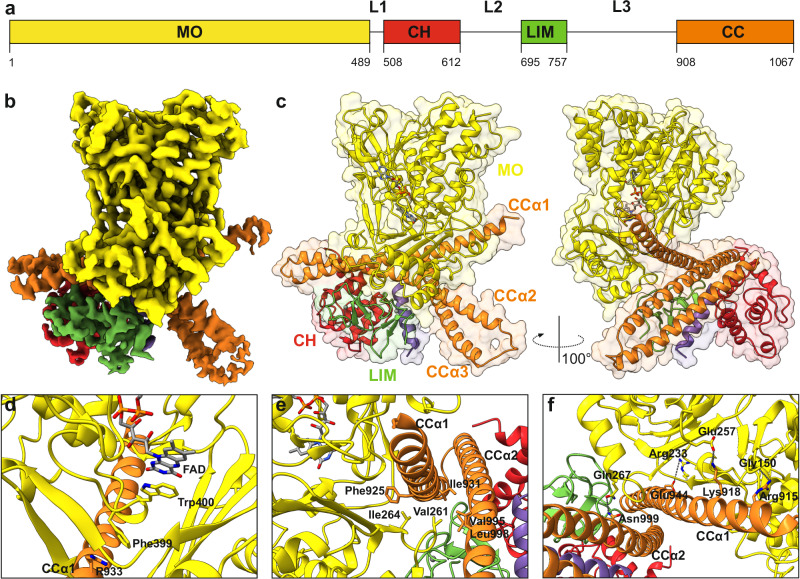
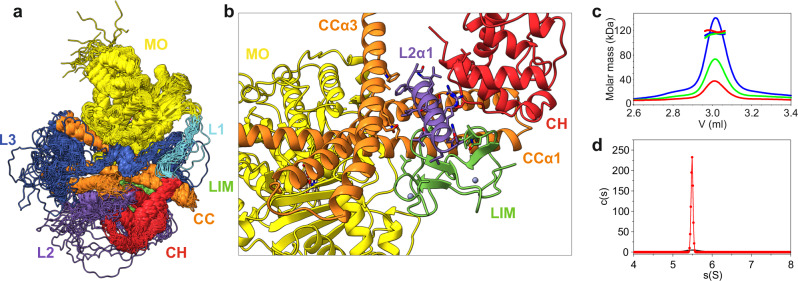
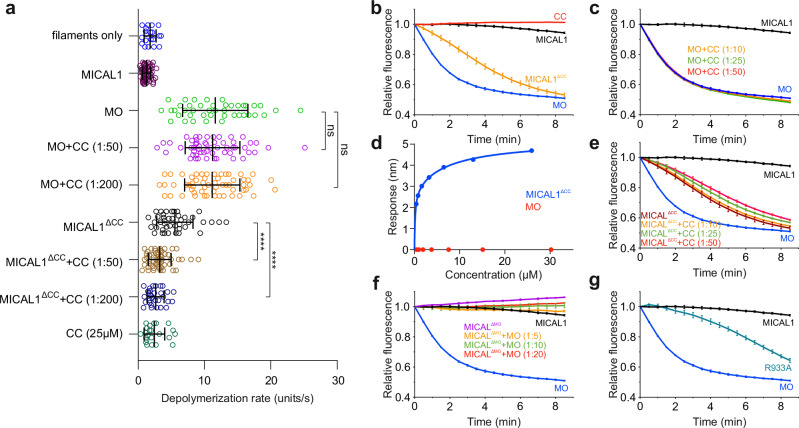
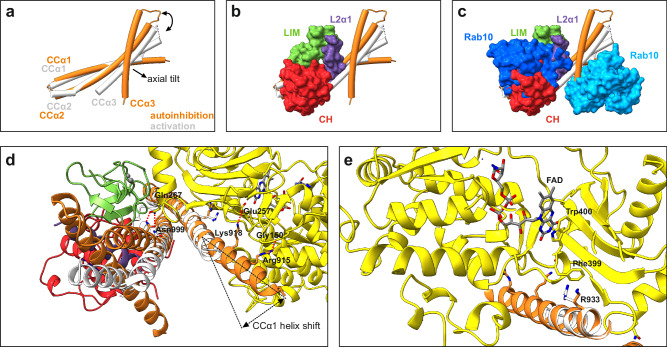
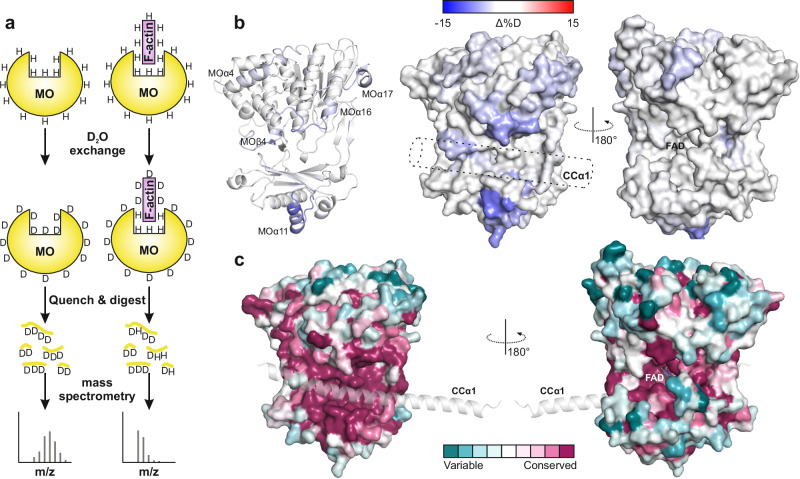
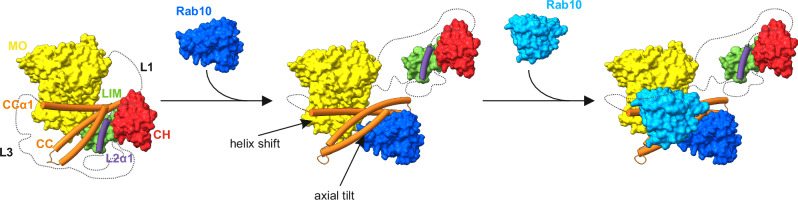
MICAL proteins play a crucial role in cellular dynamics by binding and disassembling actin filaments, impacting processes like axon guidance, cytokinesis, and cell morphology. Their cellular activity is tightly controlled, as dysregulation can lead to detrimental effects on cellular morphology. Although previous studies have suggested that MICALs are autoinhibited, and require Rab proteins to become active, the detailed molecular mechanisms remained unclear. Here, we report the cryo-EM structure of human MICAL1 at a nominal resolution of 3.1 Å. Structural analyses, alongside biochemical and functional studies, show that MICAL1 autoinhibition is mediated by an intramolecular interaction between its N-terminal catalytic and C-terminal coiled-coil domains, blocking F-actin interaction. Moreover, we demonstrate that allosteric changes in the coiled-coil domain and the binding of the tripartite assembly of CH-L2α1-LIM domains to the coiled-coil domain are crucial for MICAL activation and autoinhibition. These mechanisms appear to be evolutionarily conserved, suggesting a potential universality across the MICAL family.
© 2024. The Author(s).
Conflict of interest statement
Figures
References
-
- Terman, J. R., Mao, T., Pasterkamp, R. J., Yu, H. H. & Kolodkin, A. L. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell109, 887–900 (2002). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
