

Genetic legacy of ancient hunter-gatherer Jomon in Japanese populations
- PMID: 39532881
- PMCID: PMC11558008
- DOI: 10.1038/s41467-024-54052-0
Genetic legacy of ancient hunter-gatherer Jomon in Japanese populations
Abstract
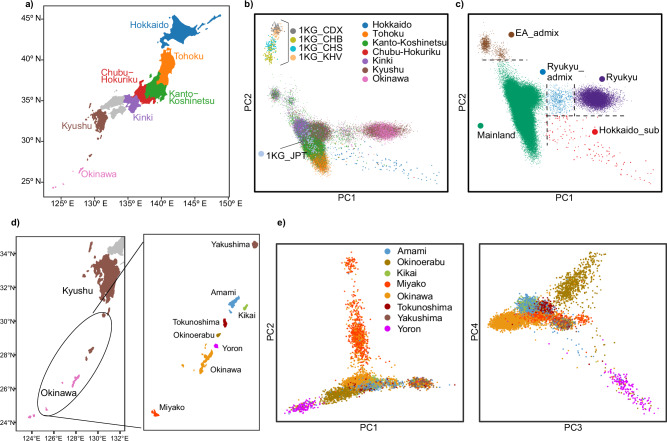
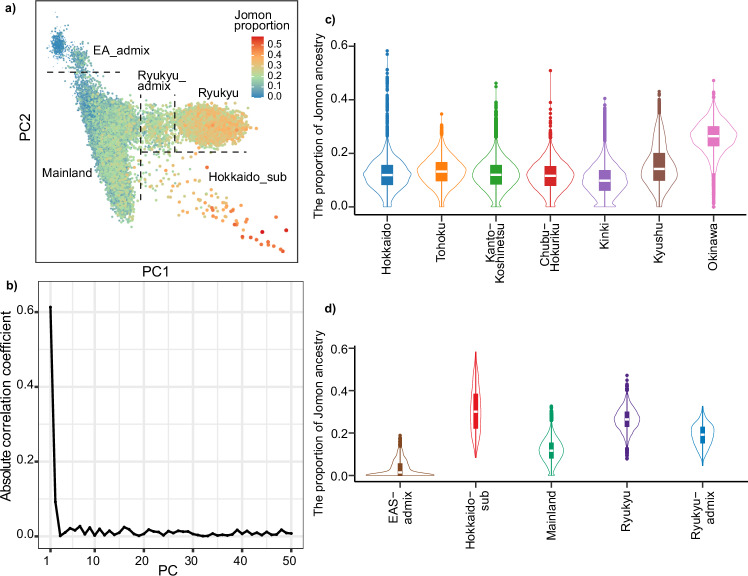
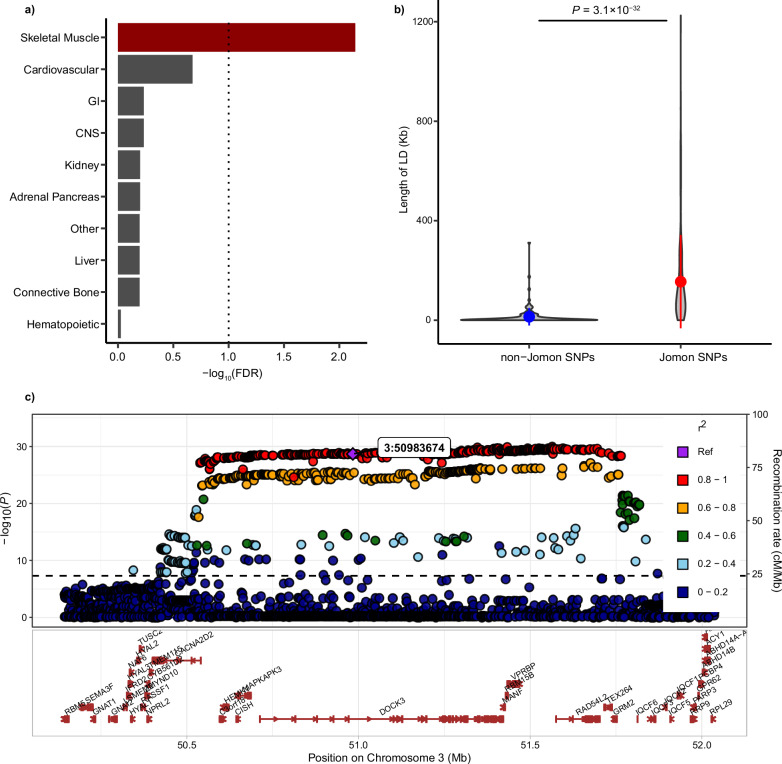
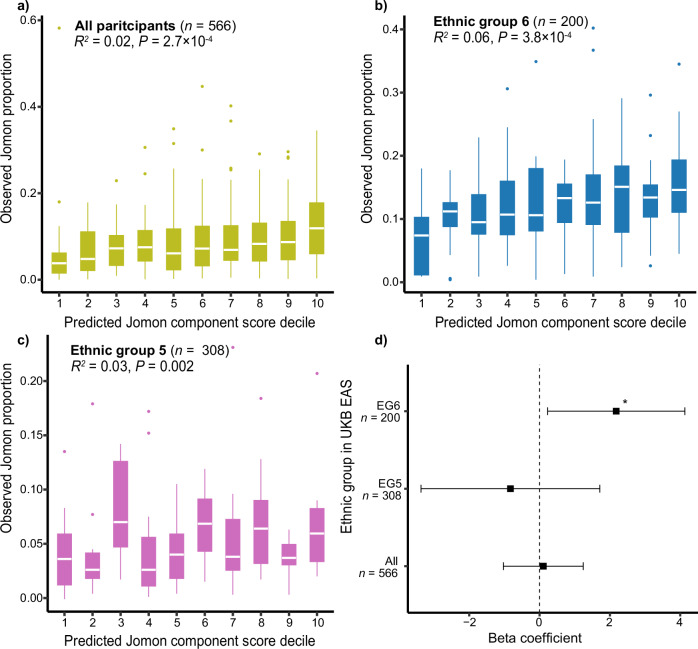
The tripartite ancestral structure is a recently proposed model for the genetic origin of modern Japanese, comprising indigenous Jomon hunter-gatherers and two additional continental ancestors from Northeast Asia and East Asia. To investigate the impact of the tripartite structure on genetic and phenotypic variation today, we conducted biobank-scale analyses by merging Biobank Japan (BBJ; n = 171,287) with ancient Japanese and Eurasian genomes (n = 22). We demonstrate the applicability of the tripartite model to Japanese populations throughout the archipelago, with an extremely strong correlation between Jomon ancestry and genomic variation among individuals. We also find that the genetic legacy of Jomon ancestry underlies an elevated body mass index (BMI). Genome-wide association analysis with rigorous adjustments for geographical and ancestral substructures identifies 132 variants that are informative for predicting individual Jomon ancestry. This prediction model is validated using independent Japanese cohorts (Nagahama cohort, n = 2993; the second cohort of BBJ, n = 72,695). We further confirm the phenotypic association between Jomon ancestry and BMI using East Asian individuals from UK Biobank (n = 2286). Our extensive analysis of ancient and modern genomes, involving over 250,000 participants, provides valuable insights into the genetic legacy of ancient hunter-gatherers in contemporary populations.
© 2024. The Author(s).
Conflict of interest statement
Figures



References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
