

Calcium signaling from damaged lysosomes induces cytoprotective stress granules
- PMID: 39533058
- PMCID: PMC11649789
- DOI: 10.1038/s44318-024-00292-1
Calcium signaling from damaged lysosomes induces cytoprotective stress granules
Abstract
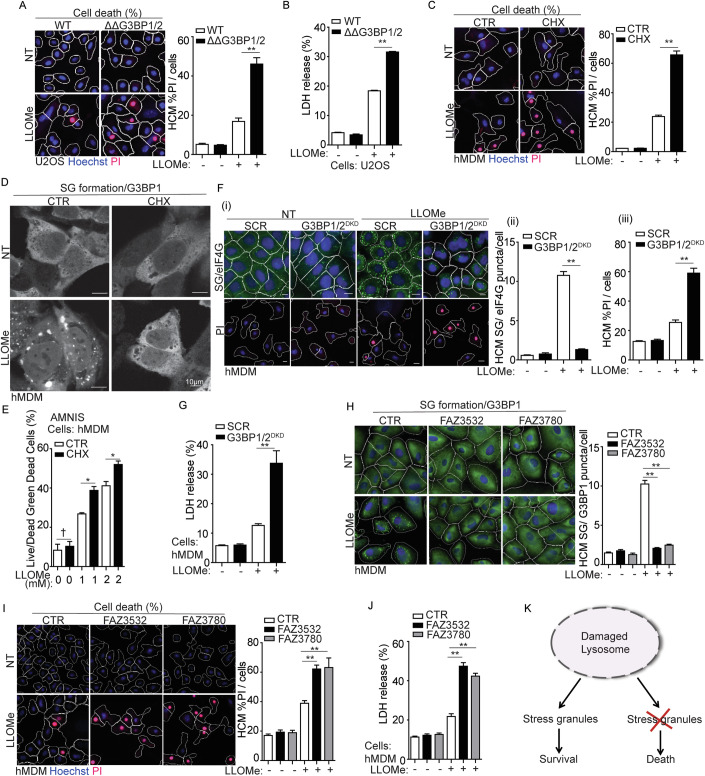
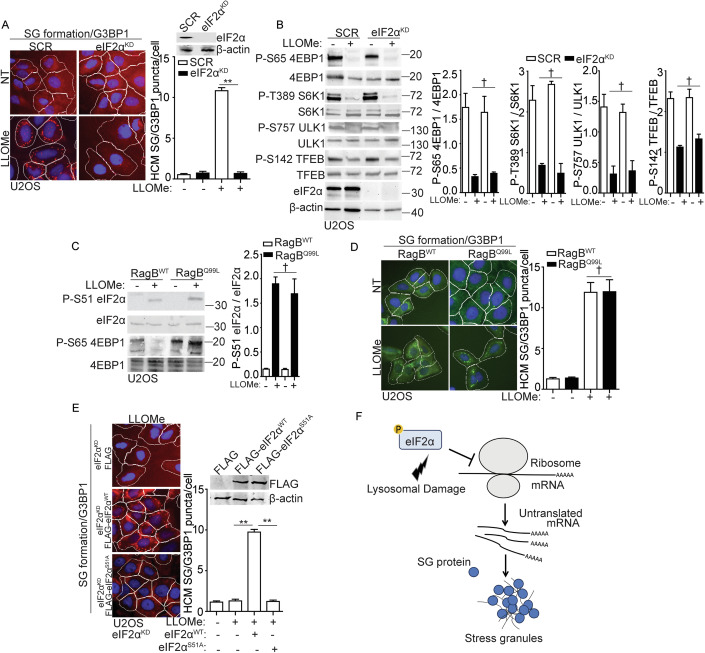
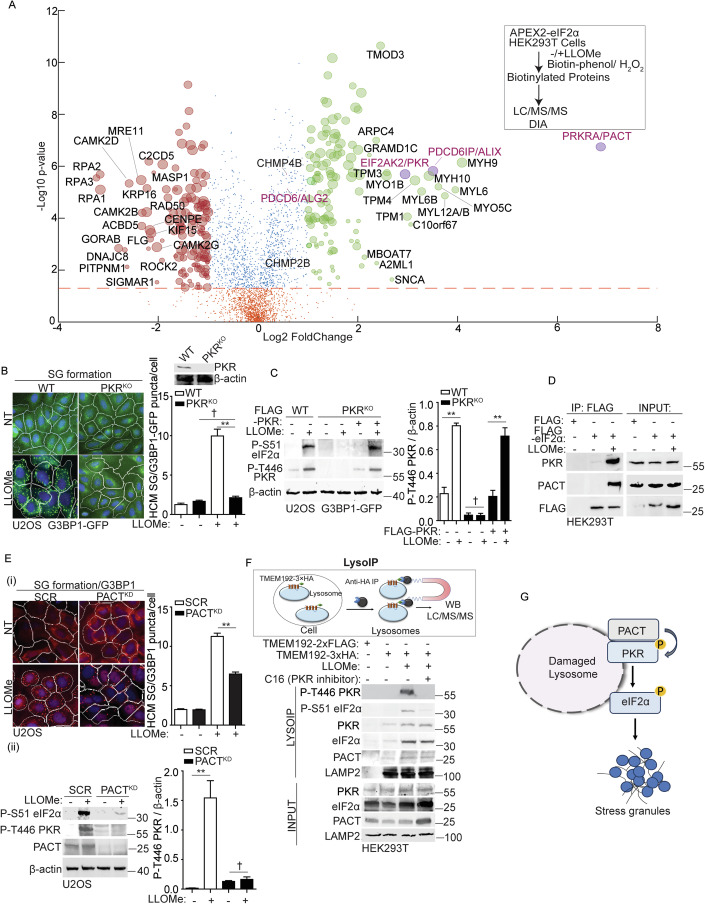
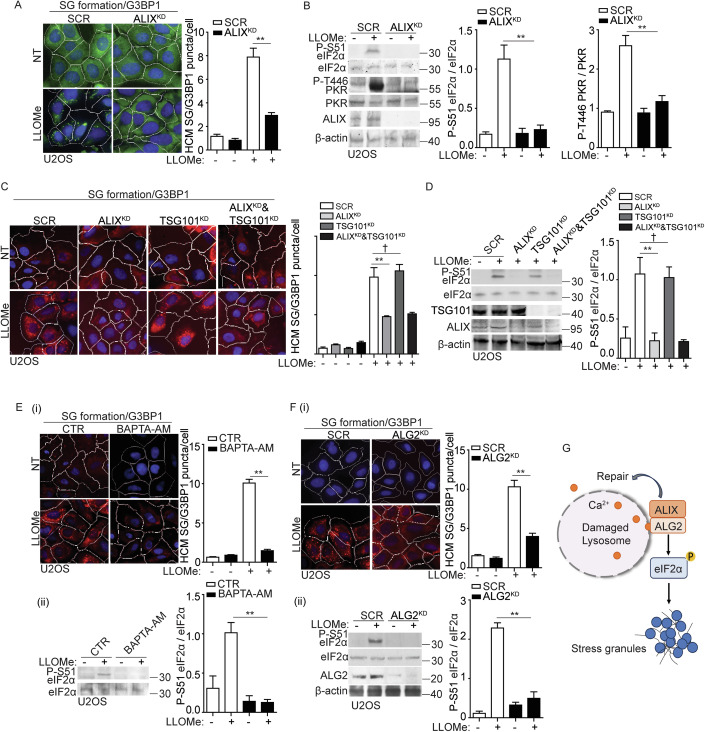
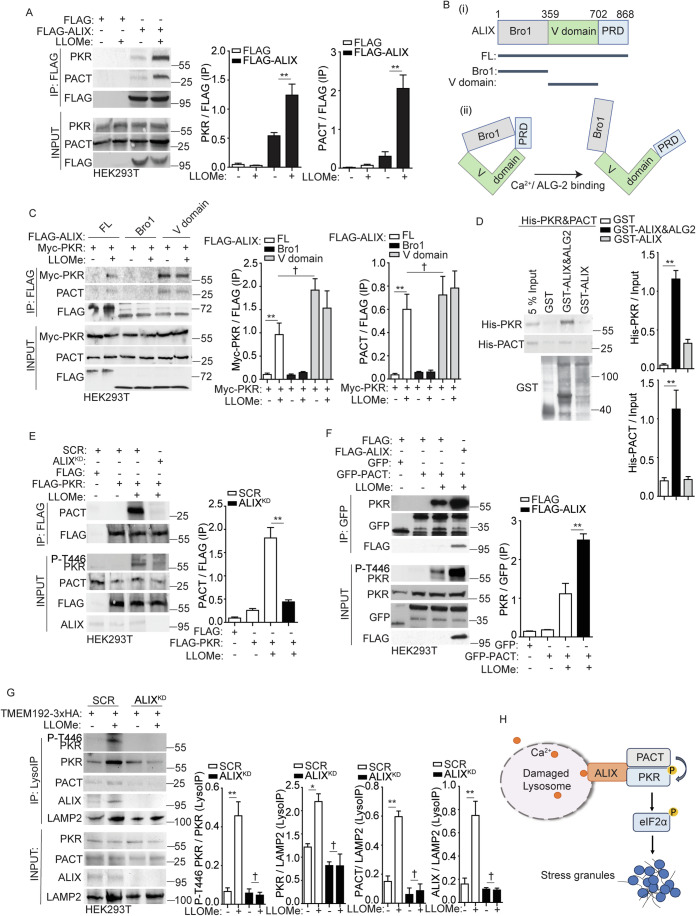
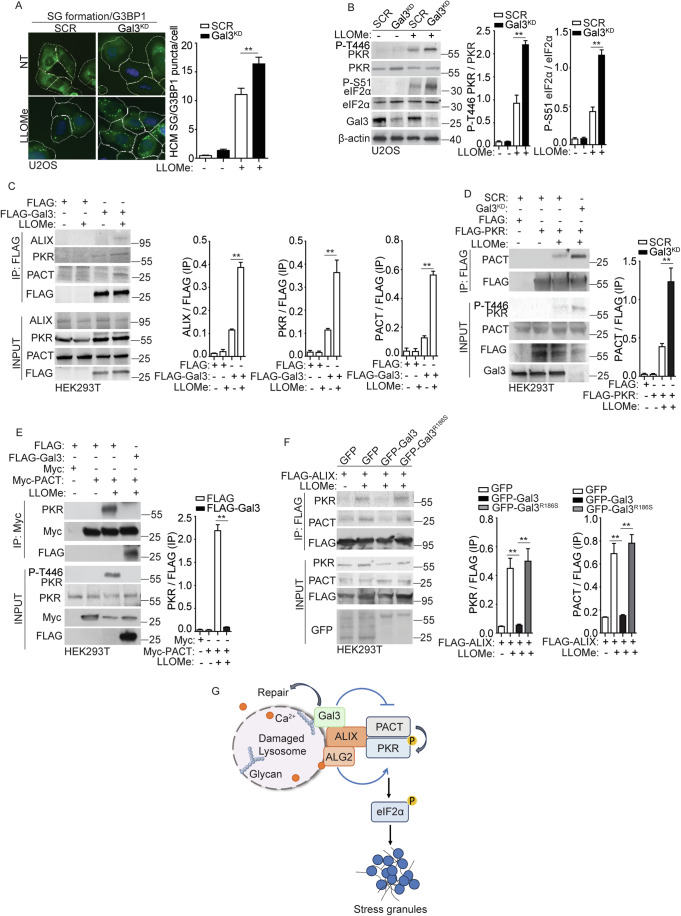
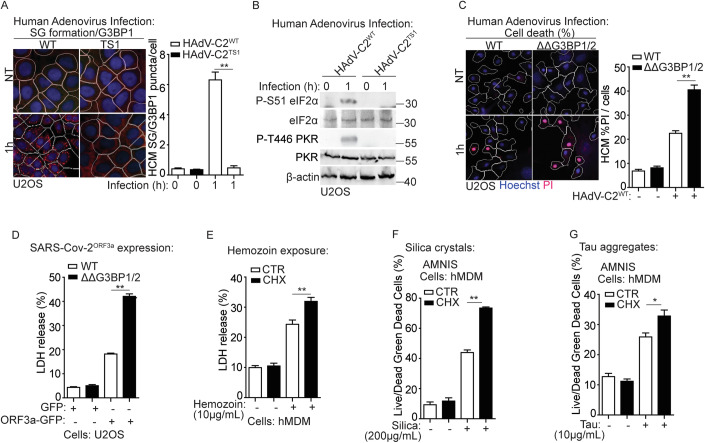
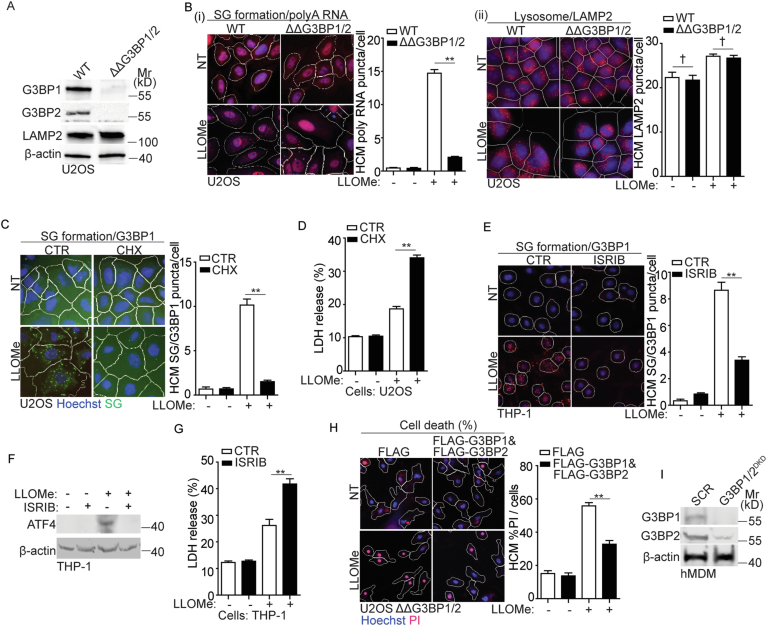
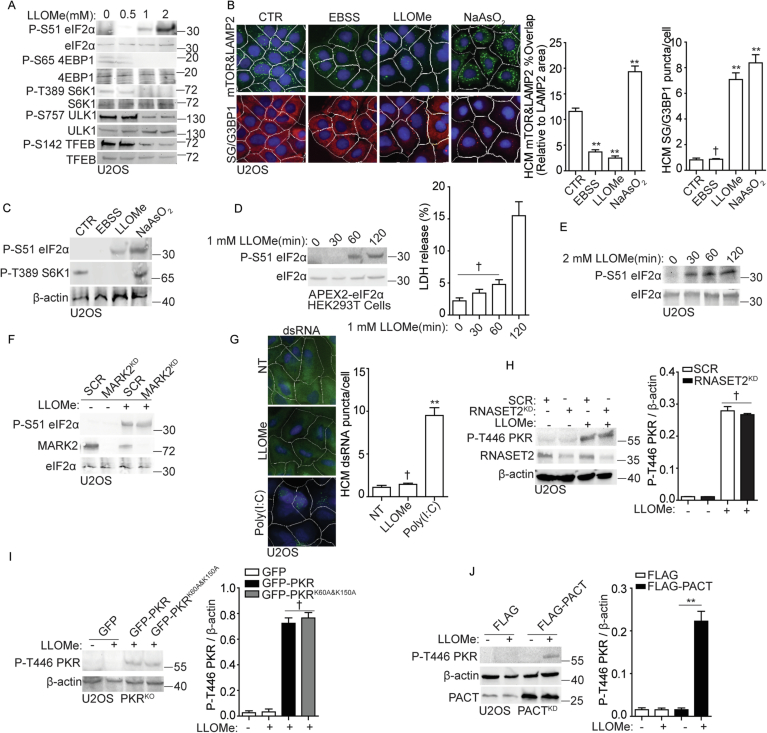
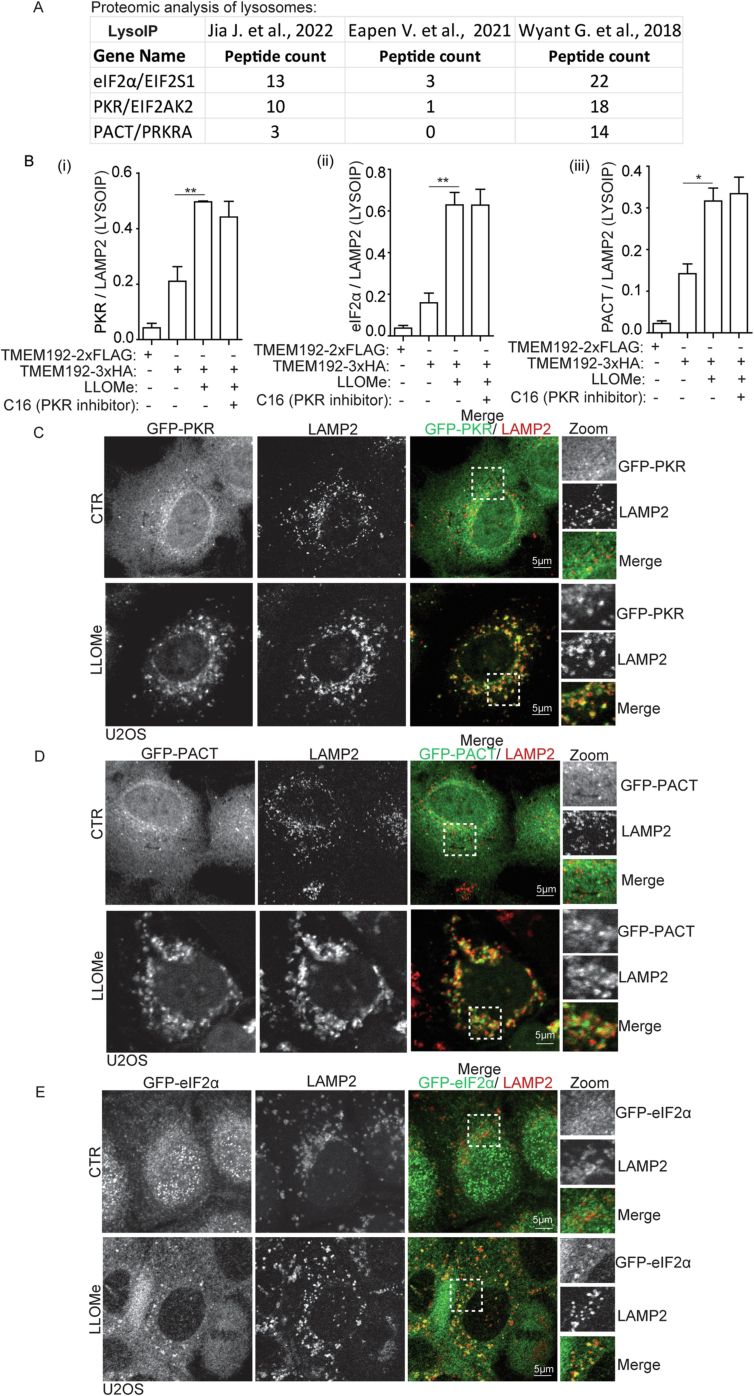
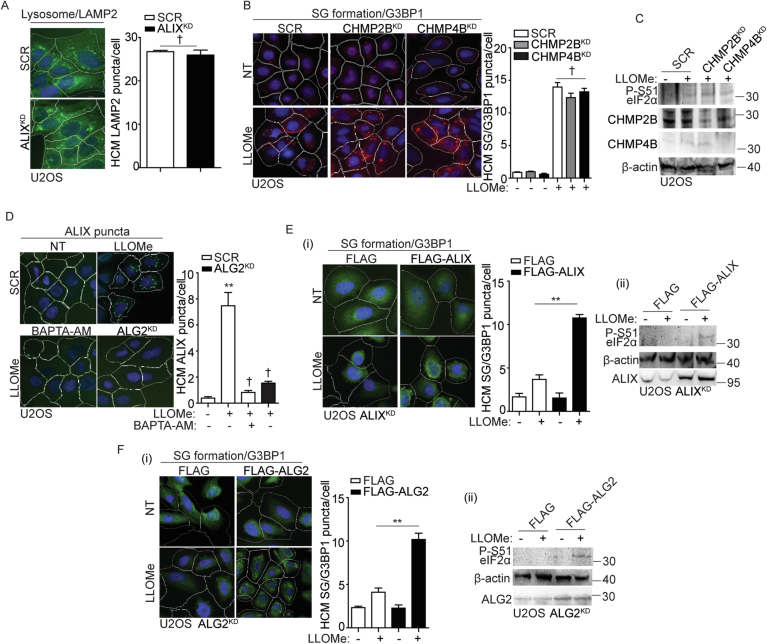
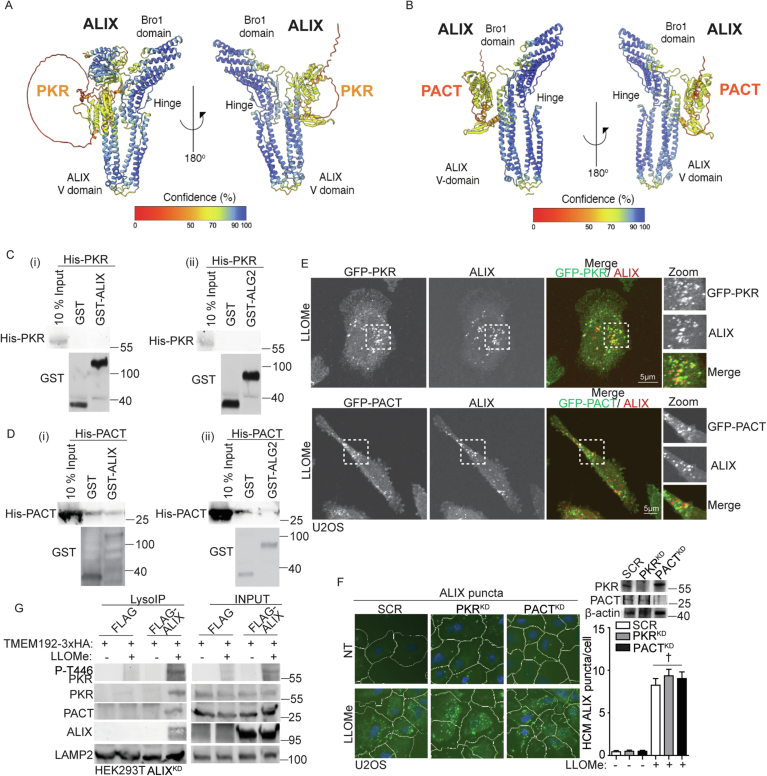
Lysosomal damage induces stress granule (SG) formation. However, the importance of SGs in determining cell fate and the precise mechanisms that mediate SG formation in response to lysosomal damage remain unclear. Here, we describe a novel calcium-dependent pathway controlling SG formation, which promotes cell survival during lysosomal damage. Mechanistically, the calcium-activated protein ALIX transduces lysosomal damage signals to SG formation by controlling eIF2α phosphorylation after sensing calcium leakage. ALIX enhances eIF2α phosphorylation by promoting the association between PKR and its activator PACT, with galectin-3 inhibiting this interaction; these regulatory events occur on damaged lysosomes. We further find that SG formation plays a crucial role in promoting cell survival upon lysosomal damage caused by factors such as SARS-CoV-2ORF3a, adenovirus, malarial pigment, proteopathic tau, or environmental hazards. Collectively, these data provide insights into the mechanism of SG formation upon lysosomal damage and implicate it in diseases associated with damaged lysosomes and SGs.
Keywords: ALG2-ALIX; Calcium-dependent Pathway; Lysosomal Damage; PACT-PKR-eIF2α; Stress Granules.
© 2024. The Author(s).
Conflict of interest statement
Disclosure and competing interests statement. The authors declare no competing interests.
Figures
References
-
- Aits S, Jäättelä M (2013) Lysosomal cell death at a glance. J Cell Sci 126(9):1905–1912 - PubMed
MeSH terms
Substances
Grants and funding
- R01EY021558 and R01EY013124/NIH RF1
- RF1NS083704/NIH RF1
- R01 EY021558/EY/NEI NIH HHS/United States
- R35GM147419/NIGMS R35
- R01 AI130473/AI/NIAID NIH HHS/United States
- P20GM121176; R35GM154651/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- P20 GM121176/GM/NIGMS NIH HHS/United States
- S10OD026918-01A1/NIH S10
- R35 GM147419/GM/NIGMS NIH HHS/United States
- S10 OD021801/OD/NIH HHS/United States
- R01 EY013124/EY/NEI NIH HHS/United States
- P20GM121176/NIH AIM COBRE
- RF1 NS083704/NS/NINDS NIH HHS/United States
- 325305/Young Research Talents Grant from the Research Council of Norway
- R35 GM154651/GM/NIGMS NIH HHS/United States
- AI130473/NIH R01
- S10 OD026918/OD/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
