

Molecular Spies in Action: Genetically Encoded Fluorescent Biosensors Light up Cellular Signals
- PMID: 39535501
- PMCID: PMC11613326
- DOI: 10.1021/acs.chemrev.4c00293
Molecular Spies in Action: Genetically Encoded Fluorescent Biosensors Light up Cellular Signals
Abstract
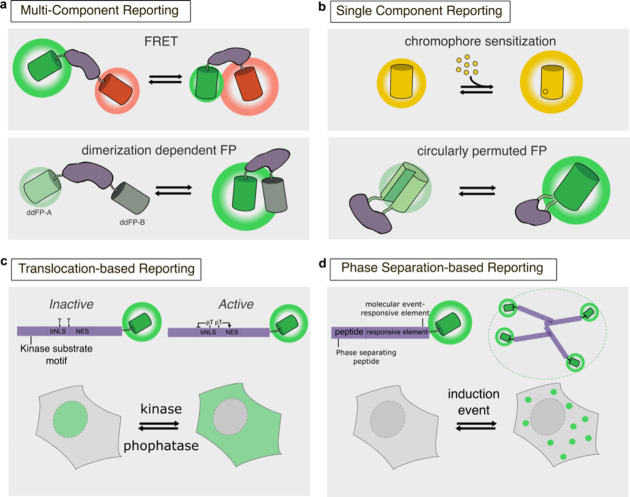
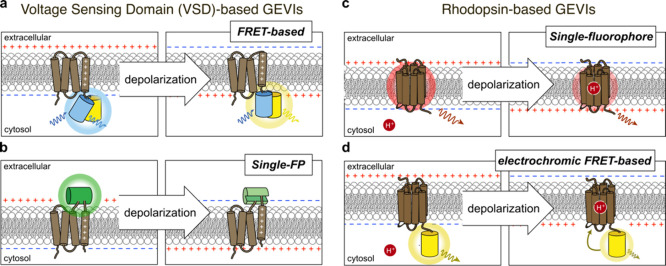
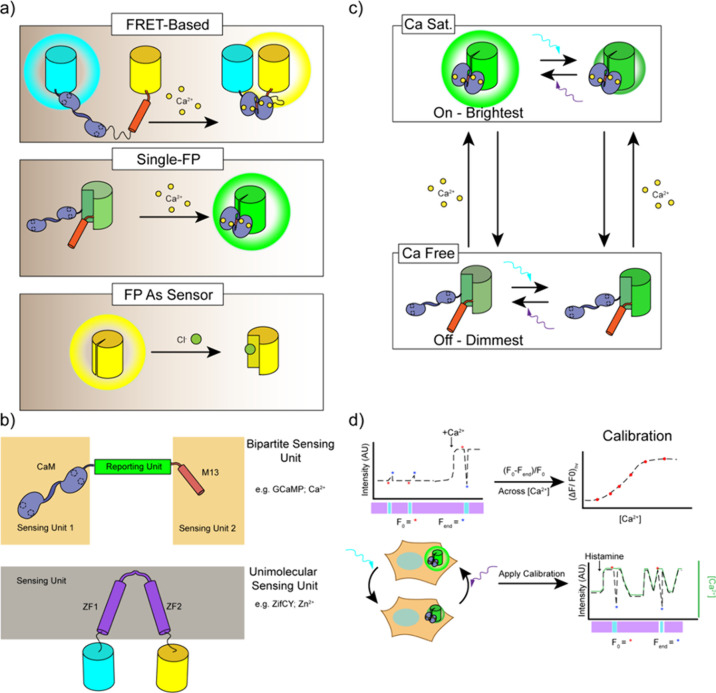
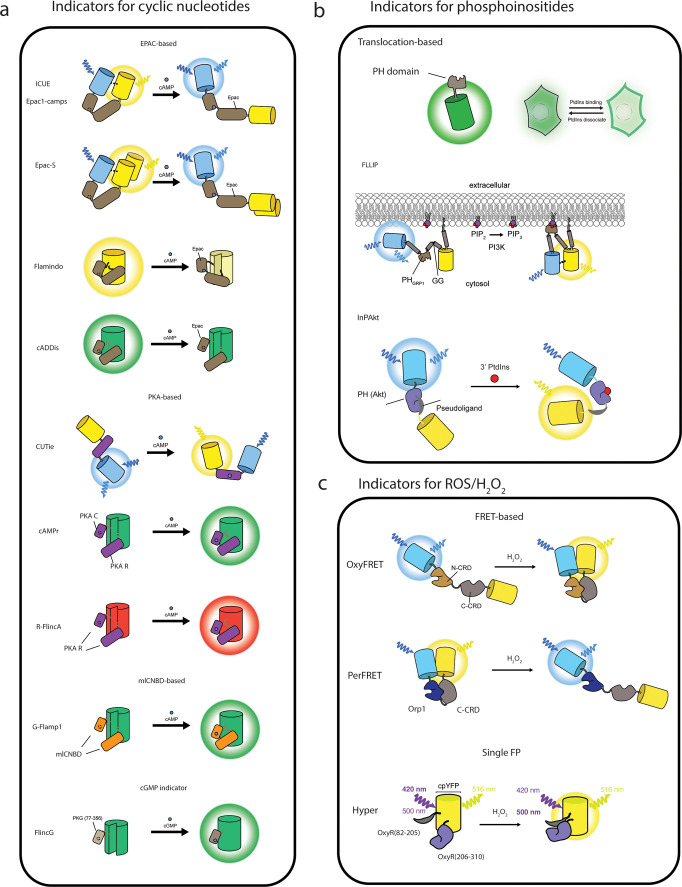
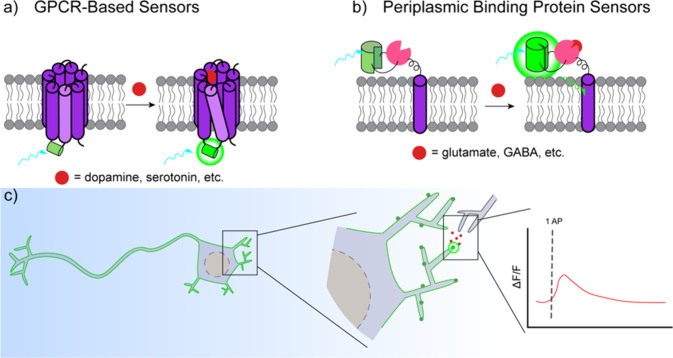
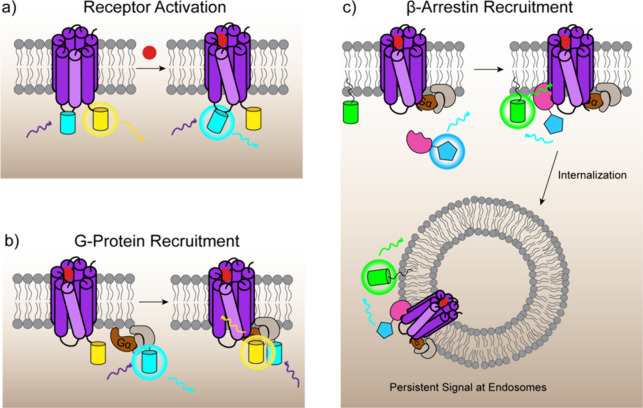
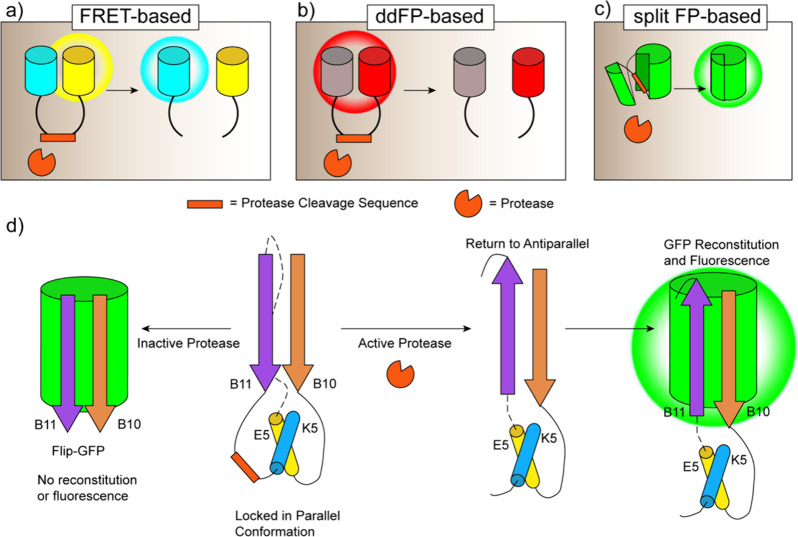
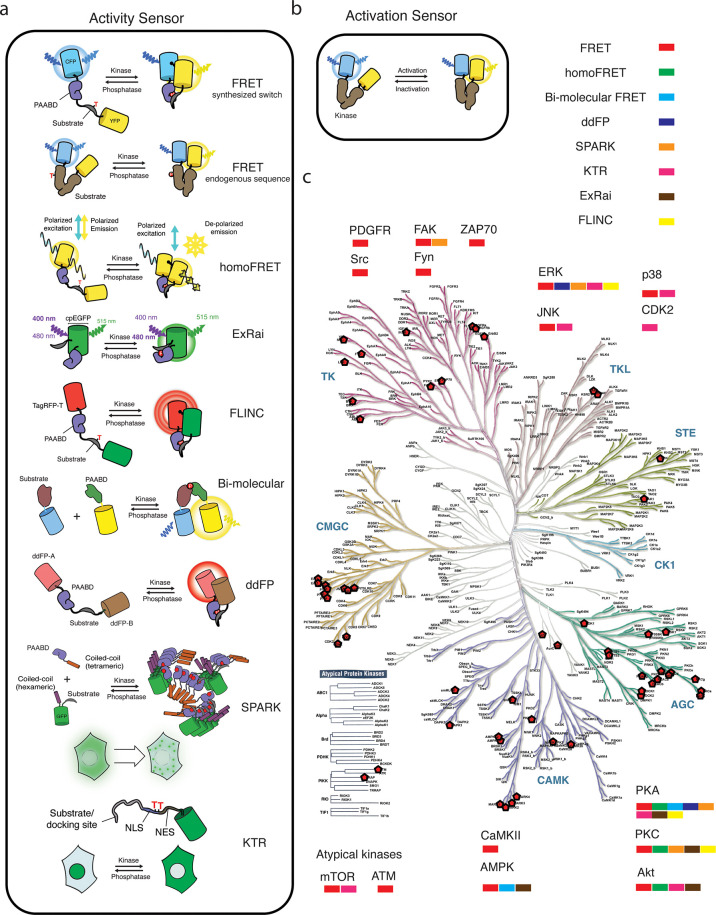
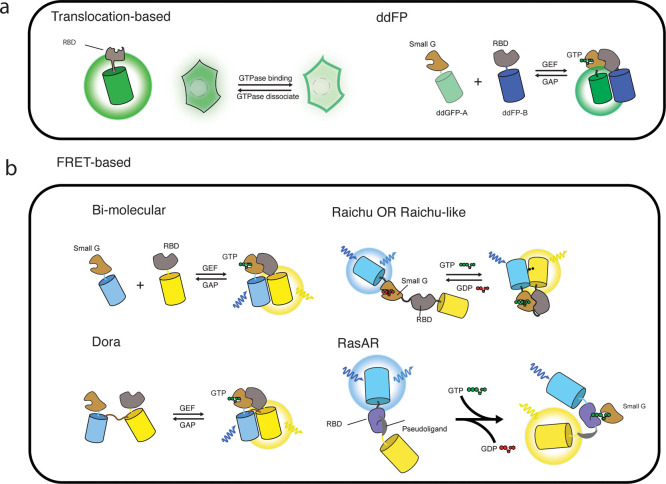
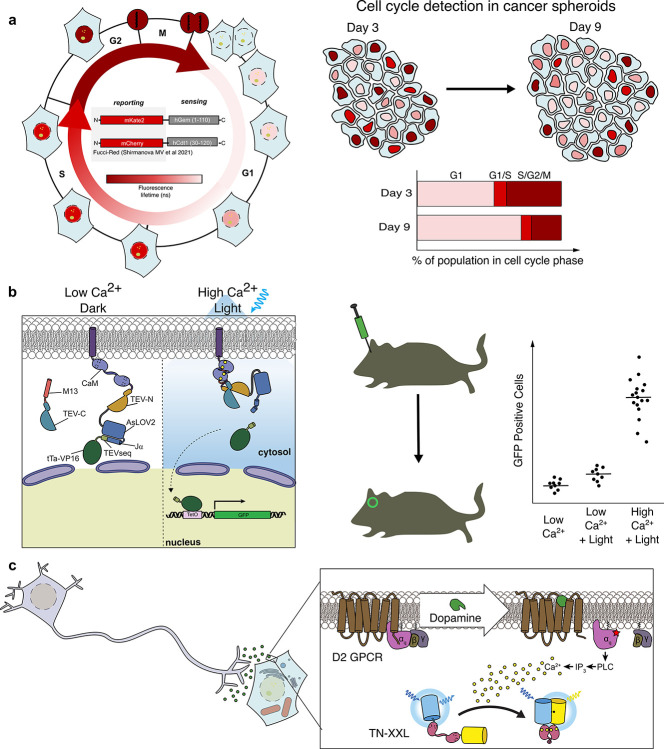
Cellular function is controlled through intricate networks of signals, which lead to the myriad pathways governing cell fate. Fluorescent biosensors have enabled the study of these signaling pathways in living systems across temporal and spatial scales. Over the years there has been an explosion in the number of fluorescent biosensors, as they have become available for numerous targets, utilized across spectral space, and suited for various imaging techniques. To guide users through this extensive biosensor landscape, we discuss critical aspects of fluorescent proteins for consideration in biosensor development, smart tagging strategies, and the historical and recent biosensors of various types, grouped by target, and with a focus on the design and recent applications of these sensors in living systems.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks.Chem Rev. 2018 Dec 26;118(24):11707-11794. doi: 10.1021/acs.chemrev.8b00333. Epub 2018 Dec 14. Chem Rev. 2018. PMID: 30550275 Free PMC article. Review.
-
Illuminating the kinome: Visualizing real-time kinase activity in biological systems using genetically encoded fluorescent protein-based biosensors.Curr Opin Chem Biol. 2020 Feb;54:63-69. doi: 10.1016/j.cbpa.2019.11.005. Epub 2020 Jan 3. Curr Opin Chem Biol. 2020. PMID: 31911398 Free PMC article. Review.
-
Design strategies of fluorescent biosensors based on biological macromolecular receptors.Sensors (Basel). 2010;10(2):1355-76. doi: 10.3390/s100201355. Epub 2010 Feb 12. Sensors (Basel). 2010. PMID: 22205872 Free PMC article. Review.
-
Live-cell imaging of cell signaling using genetically encoded fluorescent reporters.FEBS J. 2018 Jan;285(2):203-219. doi: 10.1111/febs.14134. Epub 2017 Jul 6. FEBS J. 2018. PMID: 28613457 Free PMC article. Review.
-
Genetic biosensors for imaging nitric oxide in single cells.Free Radic Biol Med. 2018 Nov 20;128:50-58. doi: 10.1016/j.freeradbiomed.2018.01.027. Epub 2018 Feb 2. Free Radic Biol Med. 2018. PMID: 29398285 Free PMC article. Review.
Cited by
-
Target-responsive vesicular nanoprobe for fluorescent detection of lipase.Mikrochim Acta. 2025 Jun 18;192(7):433. doi: 10.1007/s00604-025-07278-8. Mikrochim Acta. 2025. PMID: 40533528
-
Mechanism and application of thiol-disulfide redox biosensors with a fluorescence-lifetime readout.Proc Natl Acad Sci U S A. 2025 May 13;122(19):e2503978122. doi: 10.1073/pnas.2503978122. Epub 2025 May 6. Proc Natl Acad Sci U S A. 2025. PMID: 40327692
-
State-dependent motion of a genetically encoded fluorescent biosensor.Proc Natl Acad Sci U S A. 2025 Mar 11;122(10):e2426324122. doi: 10.1073/pnas.2426324122. Epub 2025 Mar 6. Proc Natl Acad Sci U S A. 2025. PMID: 40048274
-
High-performance genetically-encoded green and red fluorescent biosensors for pyruvate.bioRxiv [Preprint]. 2025 Jul 9:2025.04.17.649293. doi: 10.1101/2025.04.17.649293. bioRxiv. 2025. PMID: 40631294 Free PMC article. Preprint.
-
NitrOFF: An engineered fluorescent biosensor to illuminate nitrate transport in living cells.bioRxiv [Preprint]. 2025 Mar 23:2025.03.22.644677. doi: 10.1101/2025.03.22.644677. bioRxiv. 2025. PMID: 40166251 Free PMC article. Preprint.
