

Better, Faster, Stronger: Accelerating mRNA-Based Immunotherapies With Nanocarriers
- PMID: 39537215
- PMCID: PMC11655444
- DOI: 10.1002/wnan.2017
Better, Faster, Stronger: Accelerating mRNA-Based Immunotherapies With Nanocarriers
Abstract
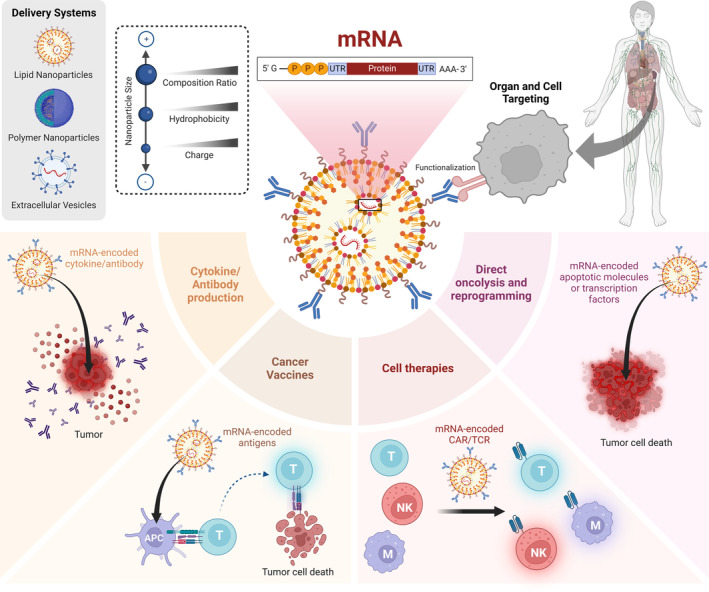
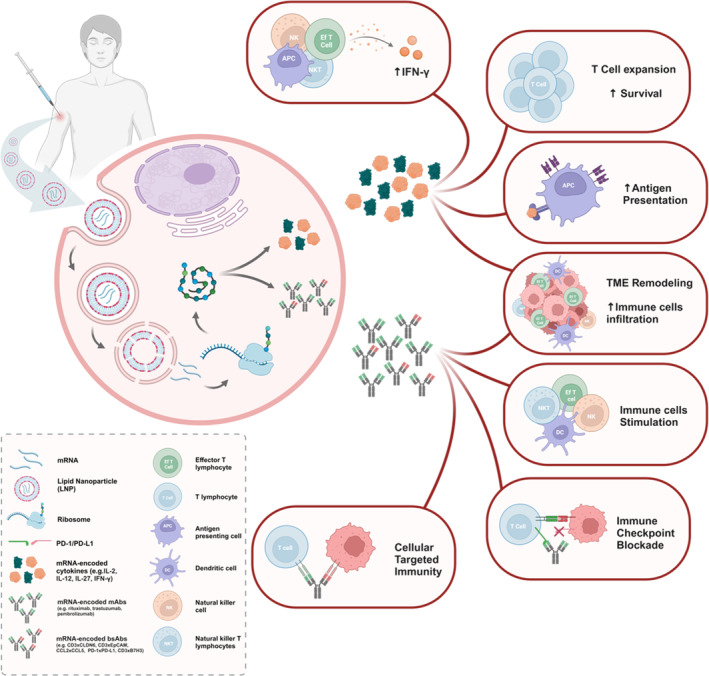
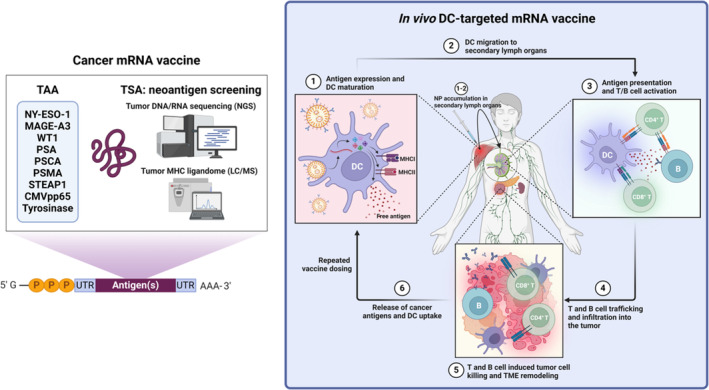
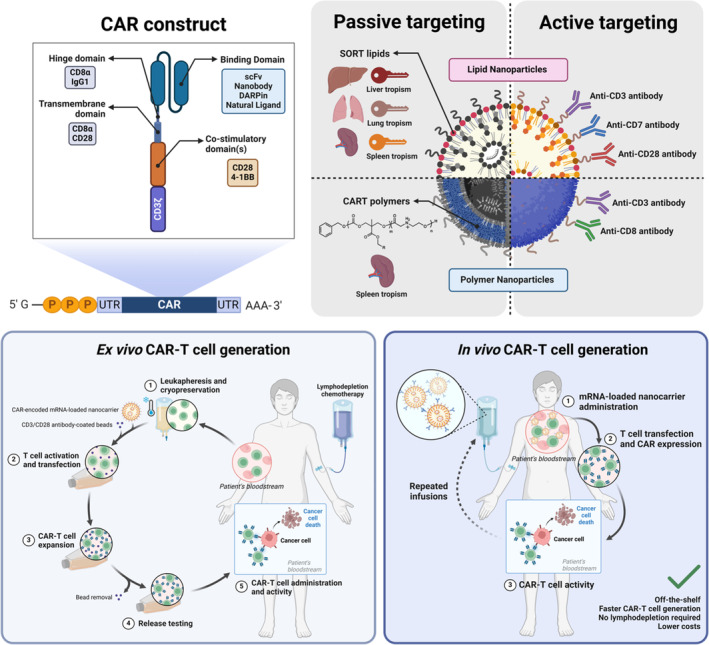
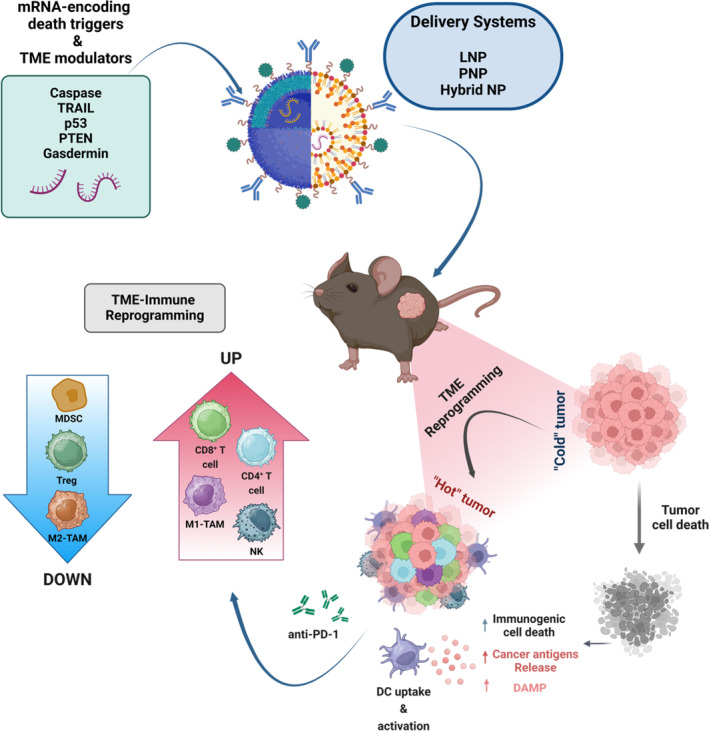
Messenger ribonucleic acid (mRNA) therapeutics are attracting attention as promising tools in cancer immunotherapy due to their ability to leverage the in vivo expression of all known protein sequences. Even small amounts of mRNA can have a powerful effect on cancer vaccines by promoting the synthesis of tumor-specific antigens (TSA) or tumor-associated antigens (TAA) by antigen-presenting cells (APC). These antigens are then presented to T cells, eliciting strong antitumor immune stimulation. The potential of mRNA can be further enhanced by expressing immunomodulatory agents, such as cytokines, antibodies, and chimeric antigen receptors (CAR), enhancing tumor immunity. Recent research also explores mRNA-encoded tumor death inducers or tumor microenvironment (TME) modulators. Despite its promise, the clinical translation of mRNA-based anticancer strategies faces challenges, including inefficient targeted delivery in vivo, failure of endosomal escape, and inadequate intracellular mRNA release, resulting in poor transfection efficiencies. Inspired by the approval of lipid nanoparticle-loaded mRNA vaccines against coronavirus disease 2019 (COVID-19) and the encouraging outcomes of mRNA-based cancer therapies in trials, innovative nonviral nanotechnology delivery systems have been engineered. These aim to advance mRNA-based cancer immunotherapies from research to clinical application. This review summarizes recent preclinical and clinical progress in lipid and polymeric nanomedicines for delivering mRNA-encoded antitumor therapeutics, including cytokines and antibody-based immunotherapies, cancer vaccines, and CAR therapies. It also addresses advanced delivery systems for direct oncolysis or TME reprogramming and highlights key challenges in translating these therapies to clinical use, exploring future perspectives, including the role of artificial intelligence and machine learning in their development.
Keywords: cancer immunotherapy; mRNA‐based delivery systems; messenger RNA (mRNA) therapeutics; nanotechnology.
© 2024 The Author(s). WIREs Nanomedicine and Nanobiotechnology published by Wiley Periodicals LLC.
Conflict of interest statement
Ronit Satchi‐Fainaro is a Board Director at Teva Pharmaceutical Industries Ltd. All the other authors declare no conflicts of interest.
Figures
Similar articles
-
Immunomodulatory nanoparticles activate cytotoxic T cells for enhancement of the effect of cancer immunotherapy.Nanoscale. 2024 Oct 3;16(38):17699-17722. doi: 10.1039/d4nr01780c. Nanoscale. 2024. PMID: 39257225 Review.
-
Lipid nanoparticle mediated mRNA delivery in cancer immunotherapy.Adv Immunol. 2025;166:37-75. doi: 10.1016/bs.ai.2025.02.001. Epub 2025 Mar 5. Adv Immunol. 2025. PMID: 40738545 Review.
-
Nanoparticles for mRNA-based cancer immunotherapy.Adv Immunol. 2025;166:77-101. doi: 10.1016/bs.ai.2024.10.012. Epub 2025 Feb 4. Adv Immunol. 2025. PMID: 40738546 Review.
-
Oncolytic virotherapy and tumor microenvironment modulation.Clin Exp Med. 2025 Jul 20;25(1):256. doi: 10.1007/s10238-025-01691-2. Clin Exp Med. 2025. PMID: 40685482 Free PMC article. Review.
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
Cited by
-
Clinical Applications of the Molecular Landscape of Melanoma: Integration of Research into Diagnostic and Therapeutic Strategies.Cancers (Basel). 2025 Apr 24;17(9):1422. doi: 10.3390/cancers17091422. Cancers (Basel). 2025. PMID: 40361349 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
- 3706/24/Israel Science Foundation
- PROF-18-682/Israel Cancer Research Fund
- LCF/PR/HR22/52420016/"la Caixa" Foundation
- LCF/PR/HR24/00968/"la Caixa" Foundation
- LCF/TR/CD20/52700005/"la Caixa" Foundation
- LCF/PR/HR19/52160021/"la Caixa" Foundation
- 615808/MRA/Melanoma Research Alliance/United States
- 591187/H2020 European Research Council
- 835227/H2020 European Research Council
- Morris Kahn Foundation
- UIDB/04138/2020/Fundação para a Ciência e Tecnologia-Ministério da Ciência, Tecnologia e Ensino Superior (FCT-MCTES)
- UIDP/04138/2020/Fundação para a Ciência e Tecnologia-Ministério da Ciência, Tecnologia e Ensino Superior (FCT-MCTES)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
