

Structural variation in the pangenome of wild and domesticated barley
- PMID: 39537924
- PMCID: PMC11655362
- DOI: 10.1038/s41586-024-08187-1
Structural variation in the pangenome of wild and domesticated barley
Abstract
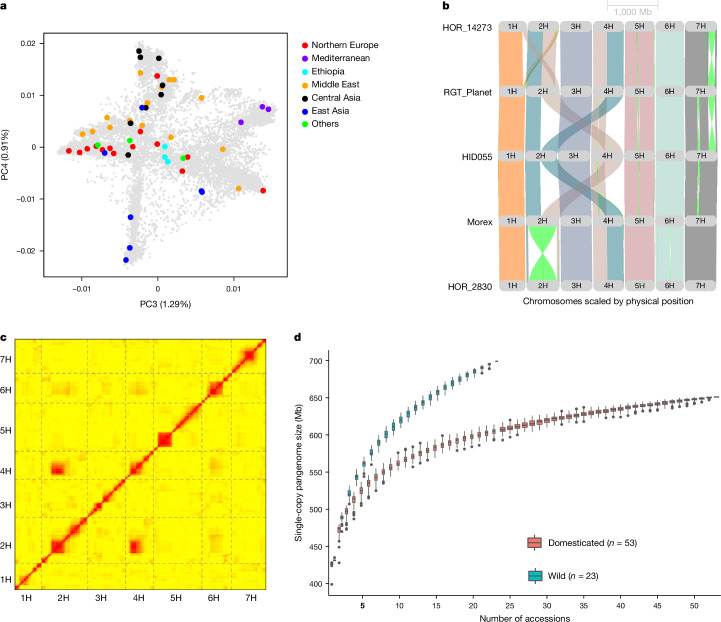
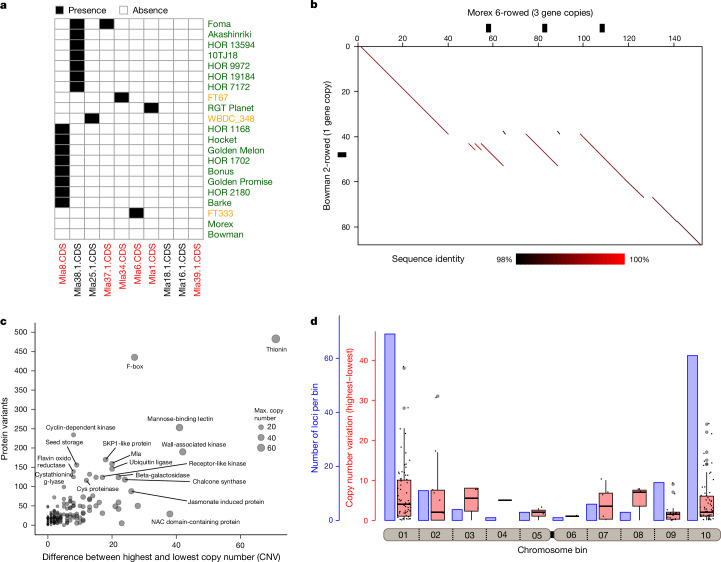
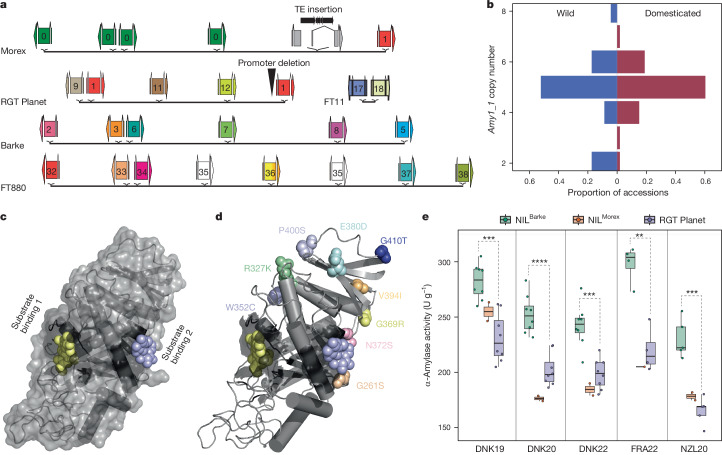
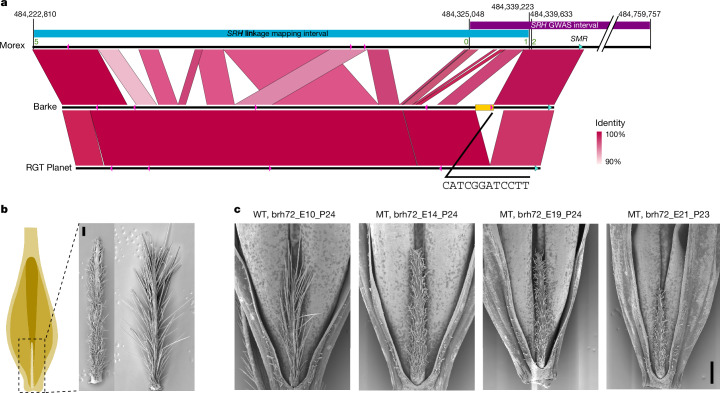
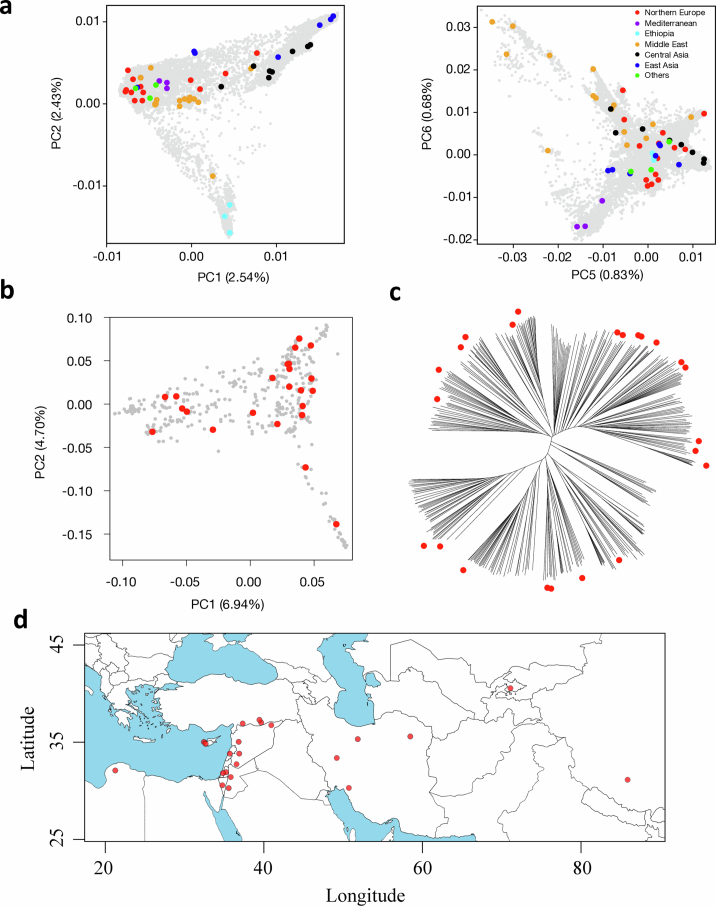
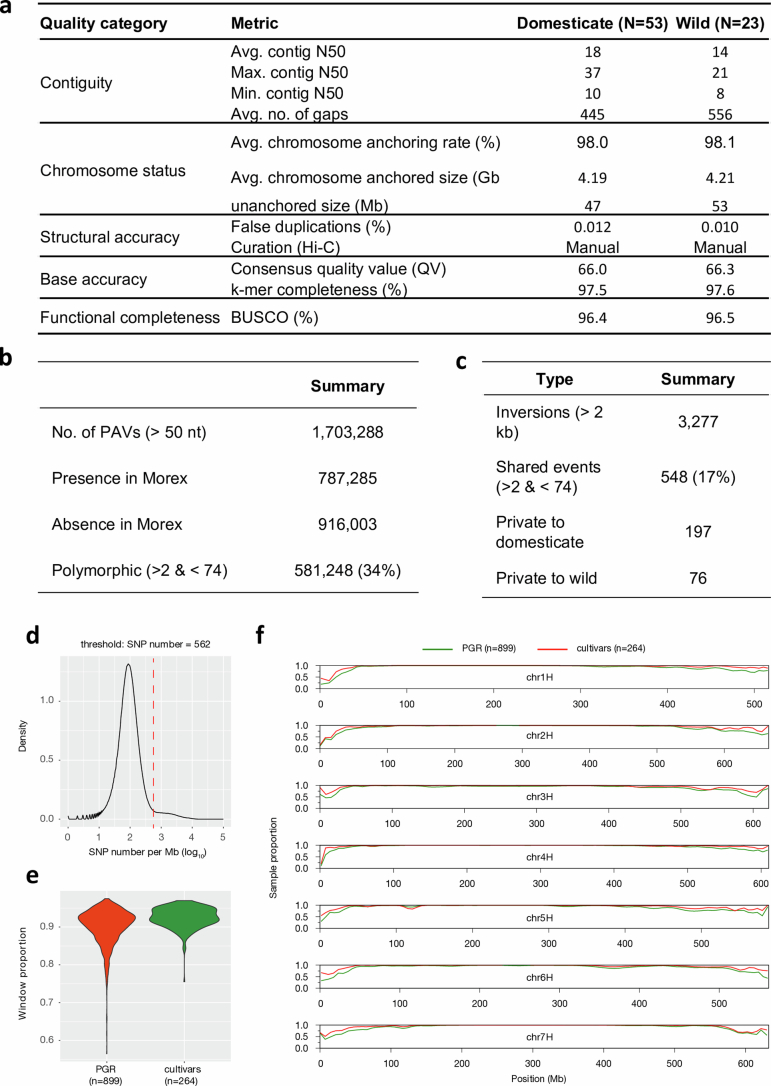
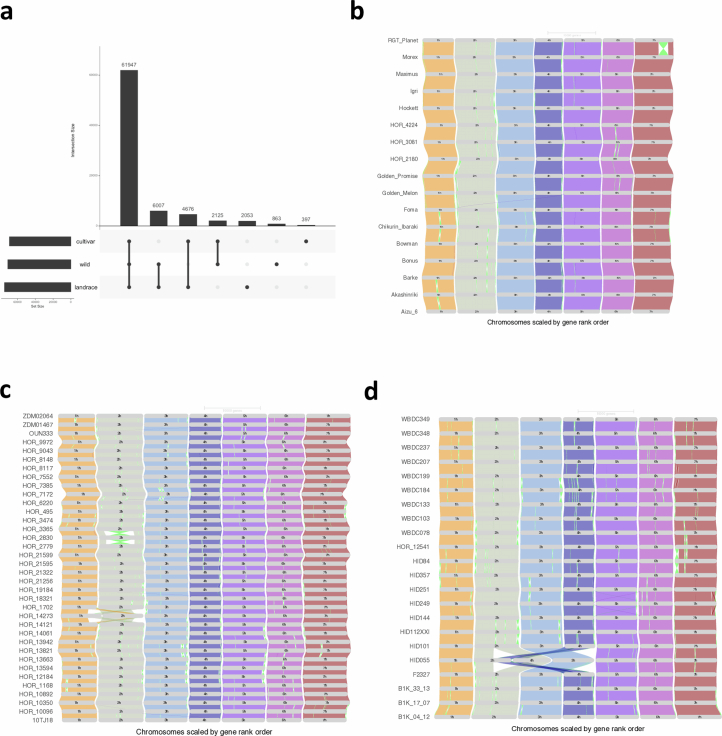
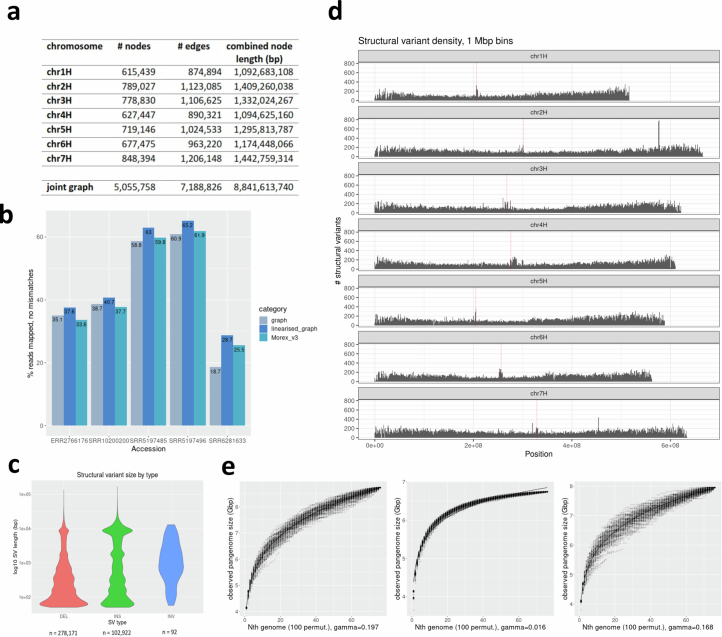
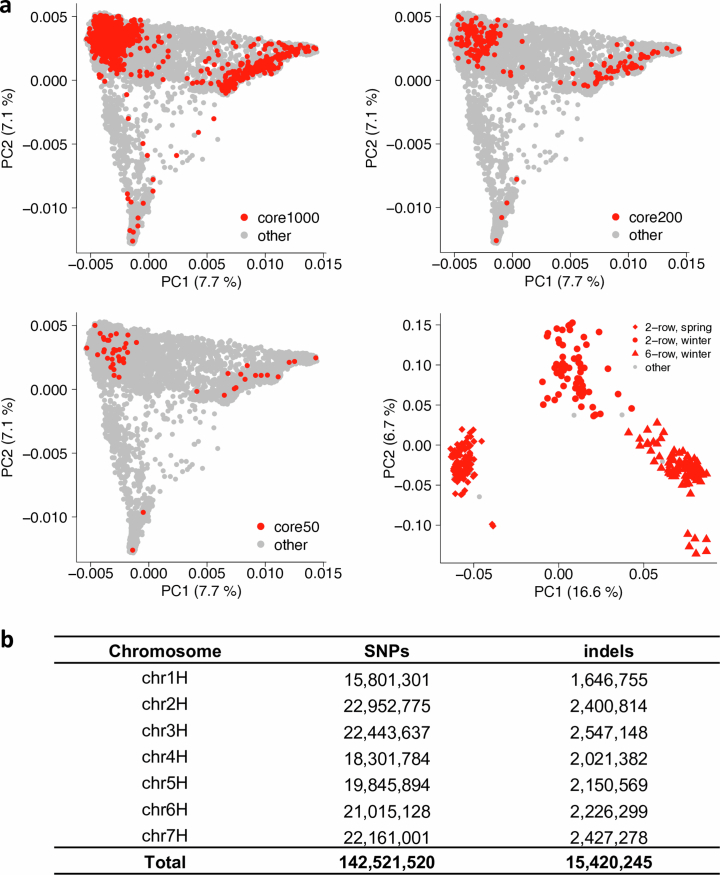
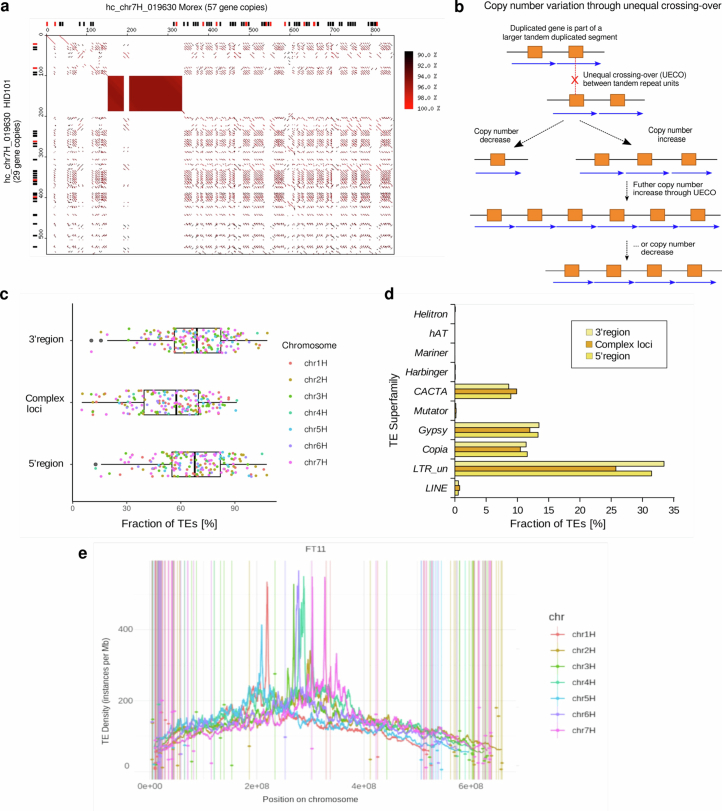
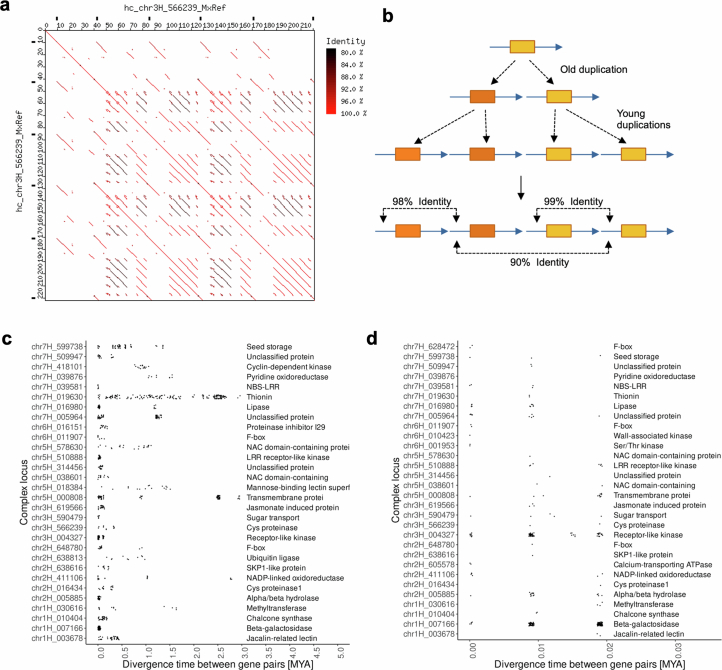
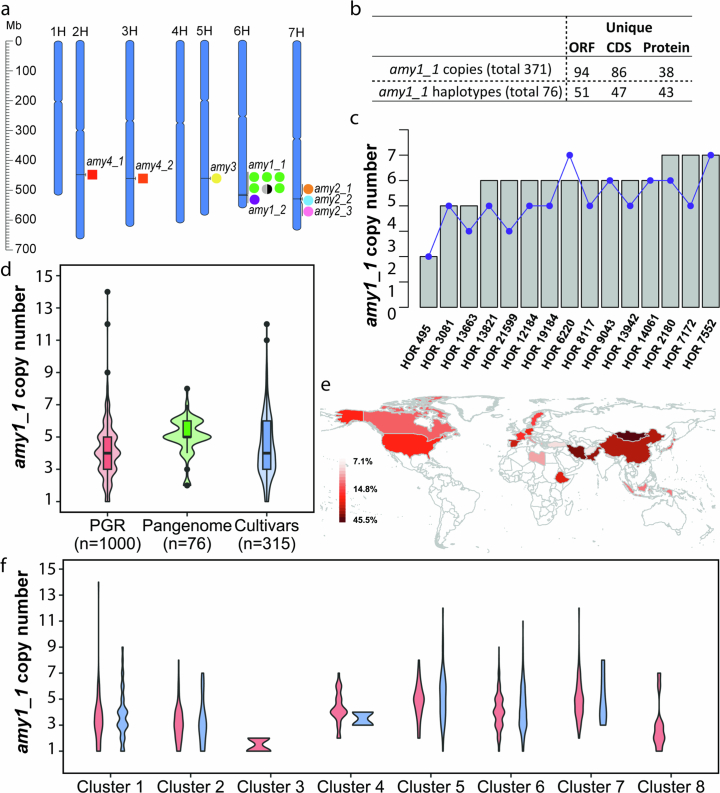
Pangenomes are collections of annotated genome sequences of multiple individuals of a species1. The structural variants uncovered by these datasets are a major asset to genetic analysis in crop plants2. Here we report a pangenome of barley comprising long-read sequence assemblies of 76 wild and domesticated genomes and short-read sequence data of 1,315 genotypes. An expanded catalogue of sequence variation in the crop includes structurally complex loci that are rich in gene copy number variation. To demonstrate the utility of the pangenome, we focus on four loci involved in disease resistance, plant architecture, nutrient release and trichome development. Novel allelic variation at a powdery mildew resistance locus and population-specific copy number gains in a regulator of vegetative branching were found. Expansion of a family of starch-cleaving enzymes in elite malting barleys was linked to shifts in enzymatic activity in micro-malting trials. Deletion of an enhancer motif is likely to change the developmental trajectory of the hairy appendages on barley grains. Our findings indicate that allelic diversity at structurally complex loci may have helped crop plants to adapt to new selective regimes in agricultural ecosystems.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: K.B., C.D., M.E.J., S.M.K., Q.L., E.M., P.R.P., B.S., H.C.T., M.T.S.N., C.V. and M.W.R. are current or previous Carlsberg A/S employees. P.A.P. and D.V. are SECOBRA Recherches employees. The other authors declare no competing interests.
Figures
References
-
- Lei, L. et al. Plant pan-genomics comes of age. Annu. Rev. Plant Biol.72, 411–435 (2021). - PubMed
-
- Sakuma, S. et al. Divergence of expression pattern contributed to neofunctionalization of duplicated HD-Zip I transcription factor in barley. New Phytol.197, 939–948 (2013). - PubMed
-
- Milner, S. G. et al. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet.51, 319–326 (2019). - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
