

A novel regimen for pancreatic ductal adenocarcinoma targeting MEK, BCL-xL, and EGFR
- PMID: 39541736
- PMCID: PMC11609319
- DOI: 10.1016/j.neo.2024.101070
A novel regimen for pancreatic ductal adenocarcinoma targeting MEK, BCL-xL, and EGFR
Abstract
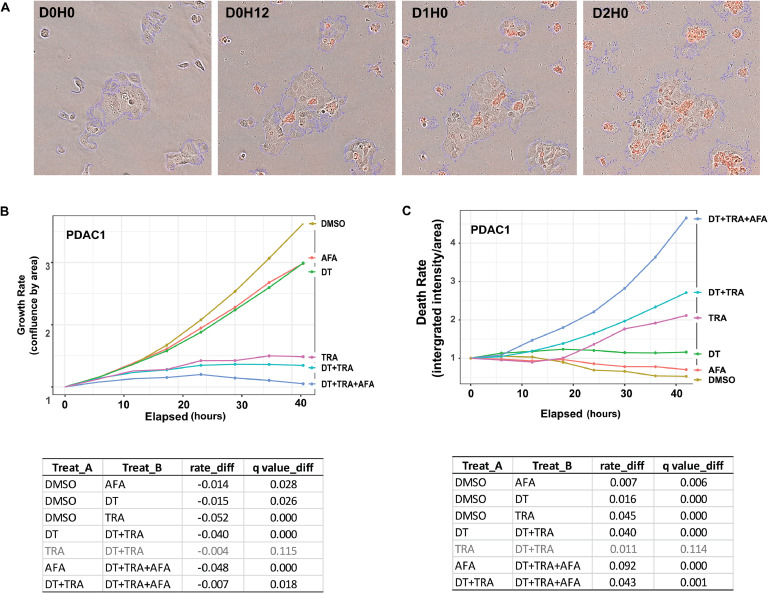
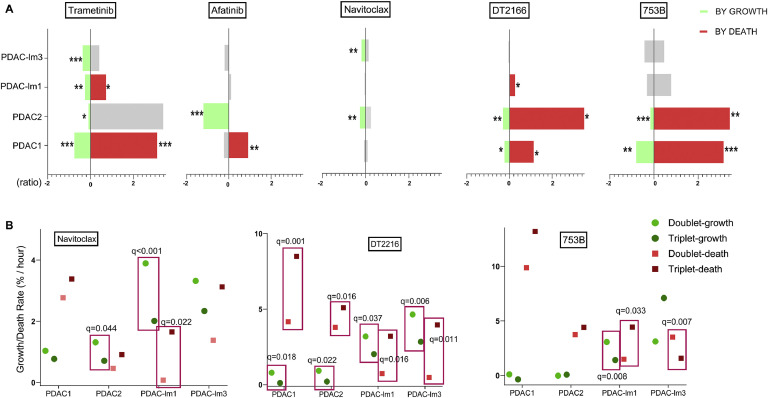
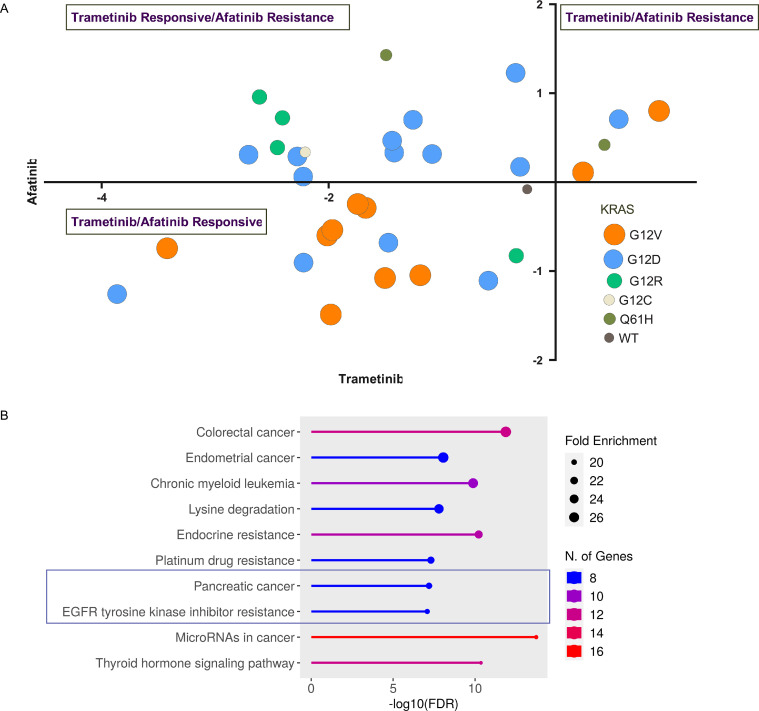
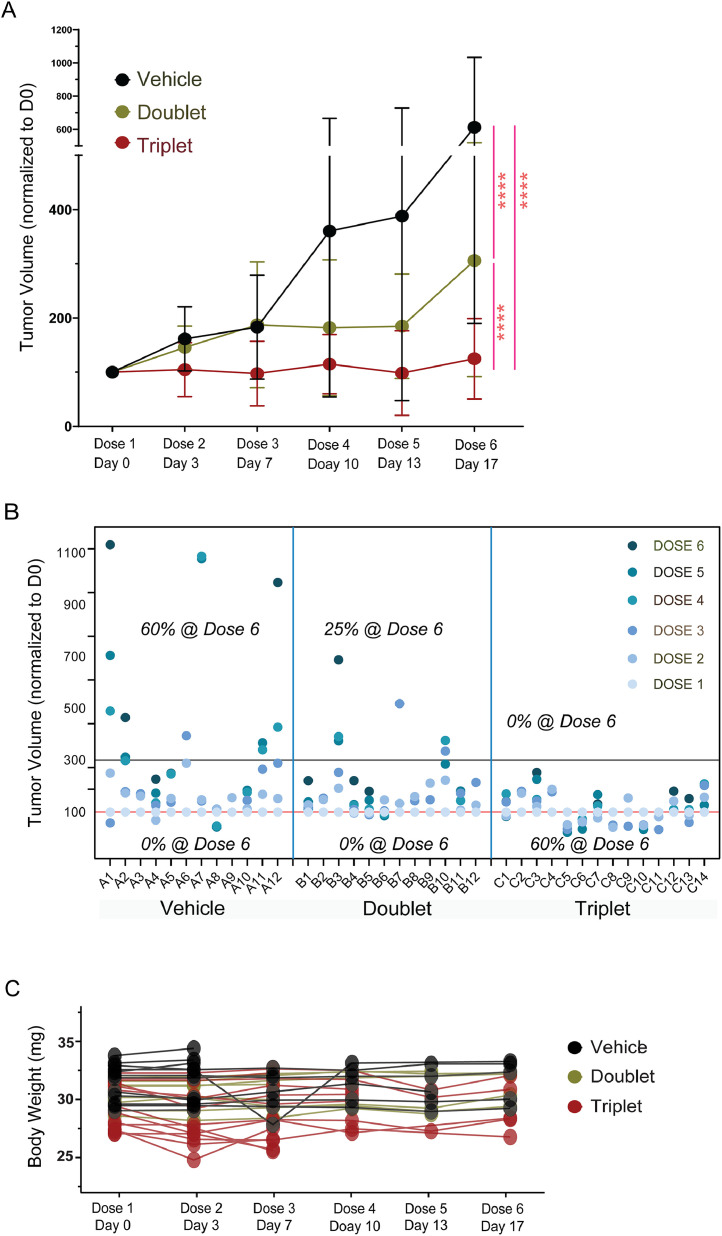
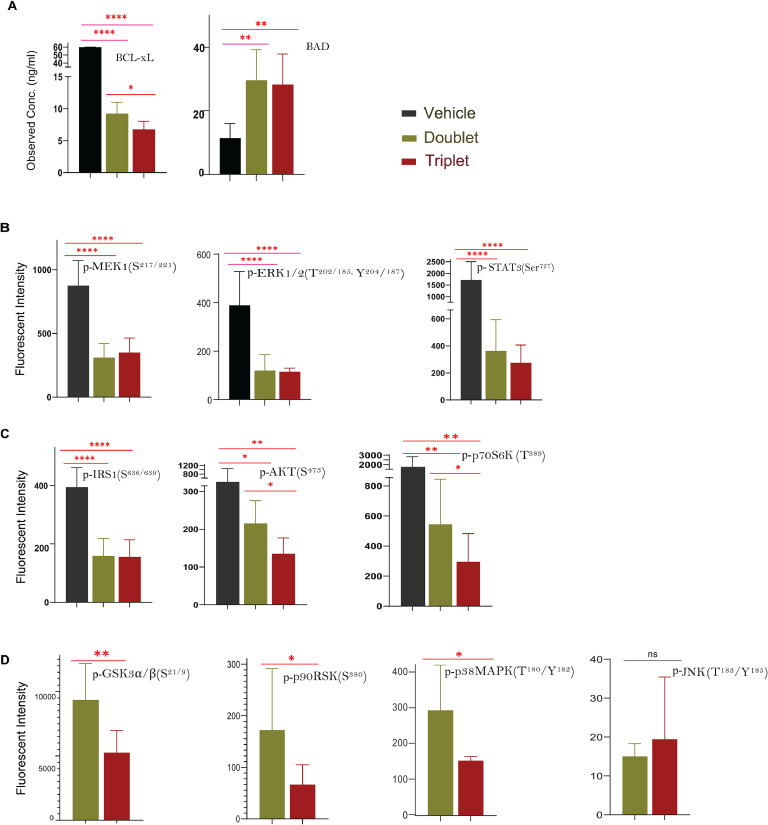
Oncogenic KRAS signaling plays a critical role in pancreatic ductal adenocarcinoma (PDAC) biology. Recent studies indicate that the combination of MEK and BCL-xL inhibition is synthetically lethal and holds promise for some types of solid cancers, however, patient response was poorly observed in PDAC predominantly due to amplified EGFR signaling. Here, we leverage the advantage of the combinational treatment strategy and designed a triplet regimen targeting the comprehensive RAS activation networks through simultaneously blocking MEK/BCL-xL/EGFR. The cytotoxicity of trametinib (MEK inhibitor), DT2216 (BCL-xL degrader) and afatinib (pan-EGFR inhibitor) and their combination was tested in patient-derived, primary PDAC cells using a live cell imaging system. Patient-derived xenograft (PDX) model was employed for the evaluation of the therapeutic efficacy and safety of the combinational regimen. Targeted pathway cascades activities were analyzed using multiplex phosphor-immune assays. In vitro comparisons showed the addition of afatinib as a third agent was statistically superior compared to a doublet of trametinib+DT2216 in suppressing cell growth and inducing cell death in all cell lines tested. This triplet similarly demonstrated significant superiority over the doublet of MEK/BCL-xL inhibition in the in vivo murine model. The triplet regimen was well tolerated in vivo. Overall tumor growth rates were significantly reduced in doublet treatment compared to controls, and further reduced in the triplet treatment group. Pathway analysis revealed the addition of afatinib in triplet regimen further inhibited PI3K/AKT effectors of p90RSK, p70S6K, and GSK3α/β along with a secondary pathway of P38 MAPK. Our study identifies an important contribution of EGFR inhibition to elevate the response of PDAC, supporting a clinical assessment of this triplet combination in patients.
Keywords: Combinational targeting therapy; EGFR, Epidermal Growth Factor Receptor; KRAS, Kirsten Rat Sarcoma Viral Oncogene Homolog; MEK, Mitogen-Activated Protein Kinase Kinase; PDAC, Pancreatic Ductal Adenocarcinoma.
Copyright © 2024. Published by Elsevier Inc.
Conflict of interest statement
Declaration of competing interest The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: P.Z., G.Z., and D.Z. are inventors of the following patent and pending patent application for use of BCL-xL PROTACs as senolytic and antitumor agents: 1. COMPOUNDS THAT INDUCE DEGRADATION OF ANTI-APOPTOTIC BCL-2 FAMILY PROTEINS AND THE USES THEREOF (Patent number: 10807977, Status: Granted, Applicant/Assignee: BioVentures, LLC, Inventors: Guangrong Zheng, Daohong Zhou, Xuan Zhang, Yingying Wang, Jianhui Chang). 2. THERAPEUTIC AGENTS AND METHODS OF TREATMENT (Patent number: 2020218367, Status: Pending, Applicant/Assignee: University of Florida Research Foundation, Inventors: Guangrong Zheng, Daohong Zhou, Pratik Pal, Xingui Liu, Dinesh Thummuri, Wenyi Hu, Peiyi Zhang, Dongwen Lyu, Yaxia Yuan, and Xuan Zhang). G.Z., and D.Z. are co-founders of and have equity in Dialectic Therapeutics, which develops BCL-xL/2 PROTACs to treat cancer. A.C. is the scientific advisory board for Dialectic that is developing DT2216 PROTAC.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
