

Digital profiling of gene expression from histology images with linearized attention
- PMID: 39543087
- PMCID: PMC11564640
- DOI: 10.1038/s41467-024-54182-5
Digital profiling of gene expression from histology images with linearized attention
Abstract
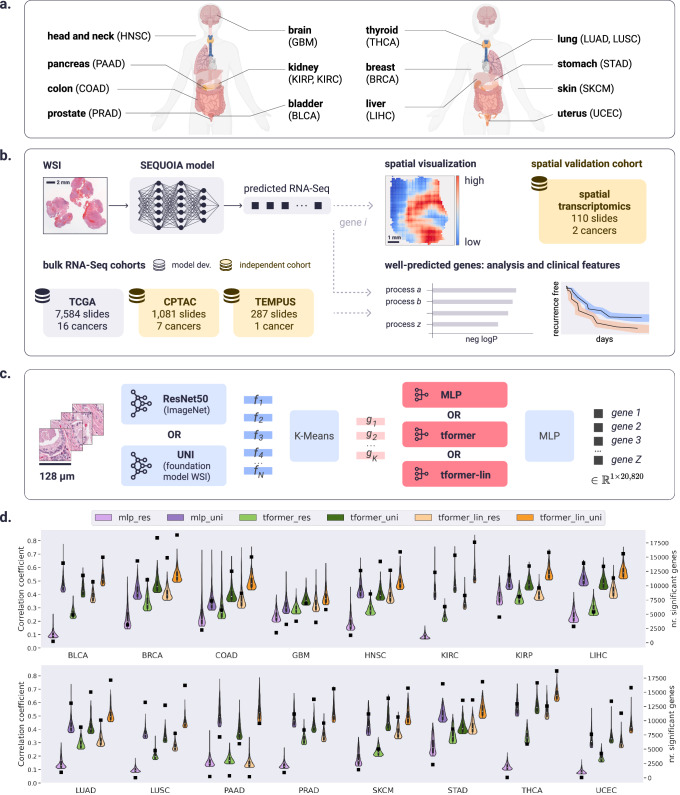
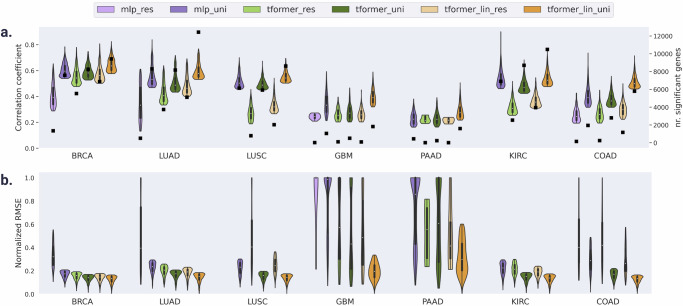
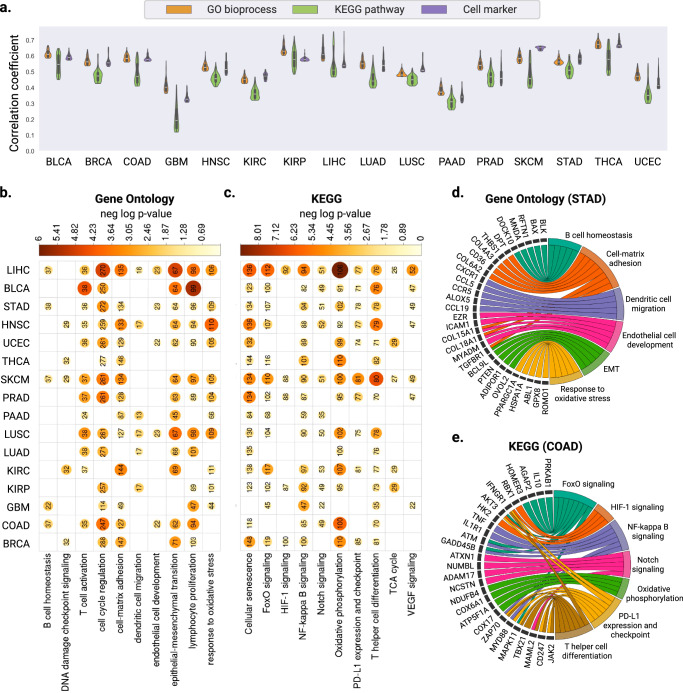
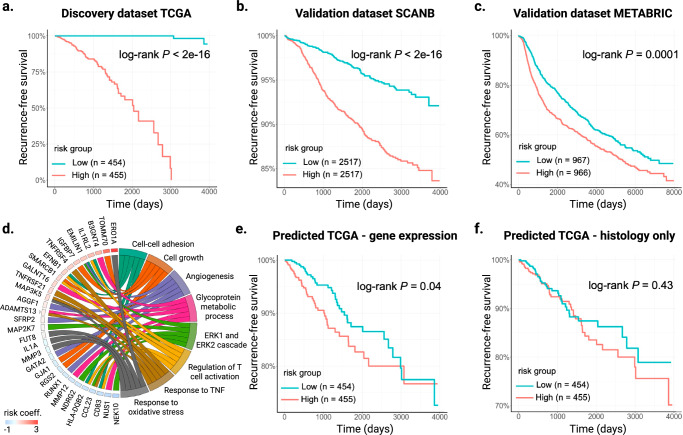
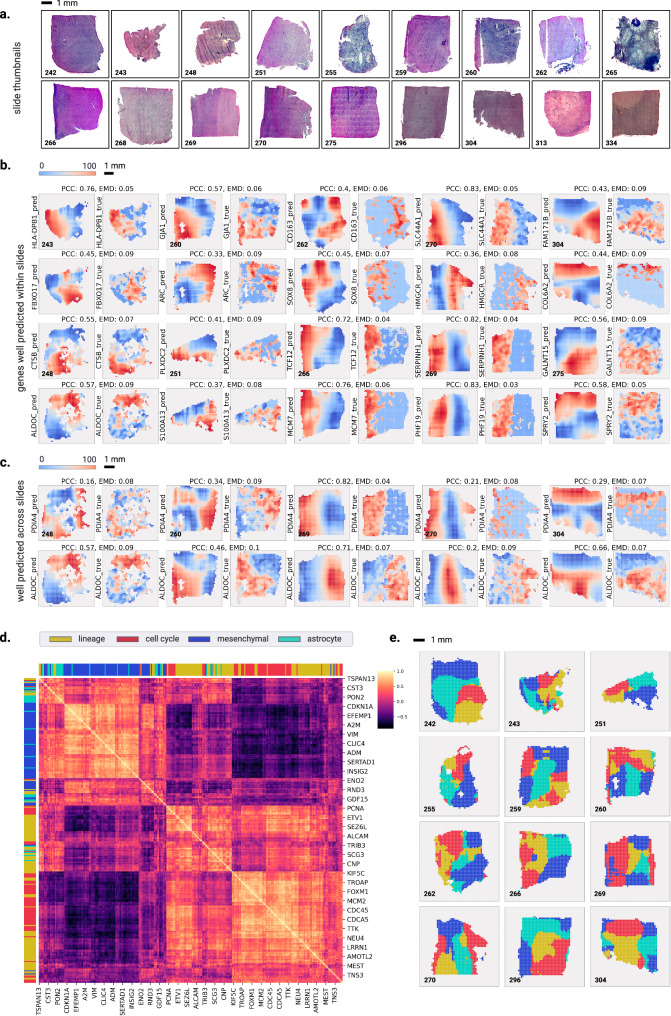
Cancer is a heterogeneous disease requiring costly genetic profiling for better understanding and management. Recent advances in deep learning have enabled cost-effective predictions of genetic alterations from whole slide images (WSIs). While transformers have driven significant progress in non-medical domains, their application to WSIs lags behind due to high model complexity and limited dataset sizes. Here, we introduce SEQUOIA, a linearized transformer model that predicts cancer transcriptomic profiles from WSIs. SEQUOIA is developed using 7584 tumor samples across 16 cancer types, with its generalization capacity validated on two independent cohorts comprising 1368 tumors. Accurately predicted genes are associated with key cancer processes, including inflammatory response, cell cycles and metabolism. Further, we demonstrate the value of SEQUOIA in stratifying the risk of breast cancer recurrence and in resolving spatial gene expression at loco-regional levels. SEQUOIA hence deciphers clinically relevant information from WSIs, opening avenues for personalized cancer management.
© 2024. The Author(s).
Conflict of interest statement
Figures
Update of
-
Digital profiling of cancer transcriptomes from histology images with grouped vision attention.bioRxiv [Preprint]. 2024 Jan 19:2023.09.28.560068. doi: 10.1101/2023.09.28.560068. bioRxiv. 2024. Update in: Nat Commun. 2024 Nov 14;15(1):9886. doi: 10.1038/s41467-024-54182-5. PMID: 37808782 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
