

Virion morphology and on-virus spike protein structures of diverse SARS-CoV-2 variants
- PMID: 39543395
- PMCID: PMC11649927
- DOI: 10.1038/s44318-024-00303-1
Virion morphology and on-virus spike protein structures of diverse SARS-CoV-2 variants
Abstract
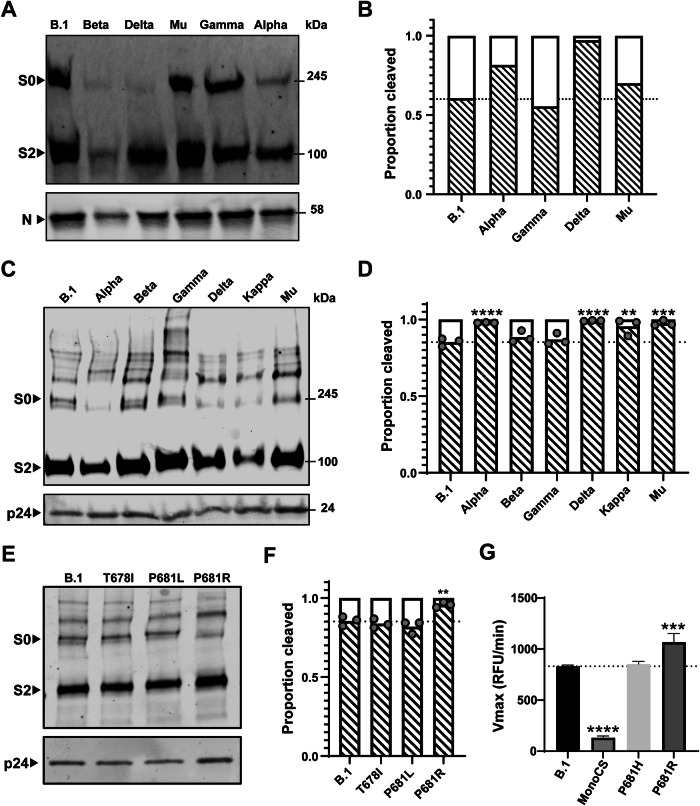
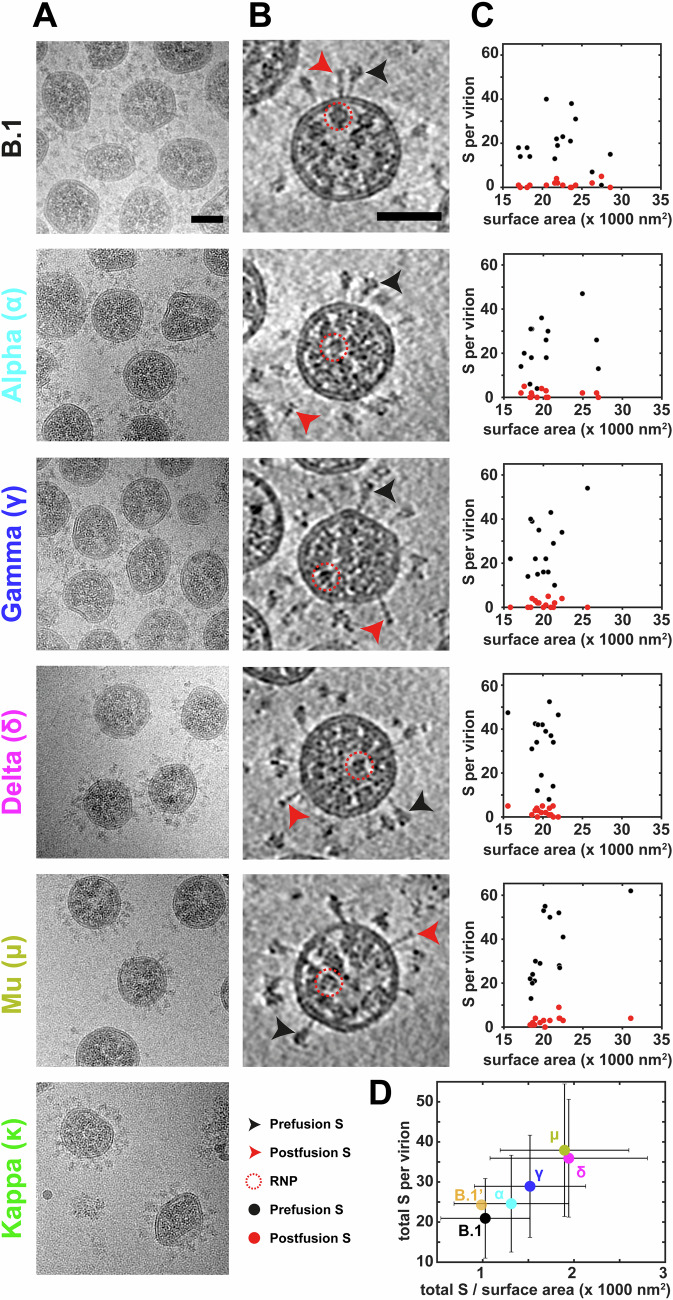
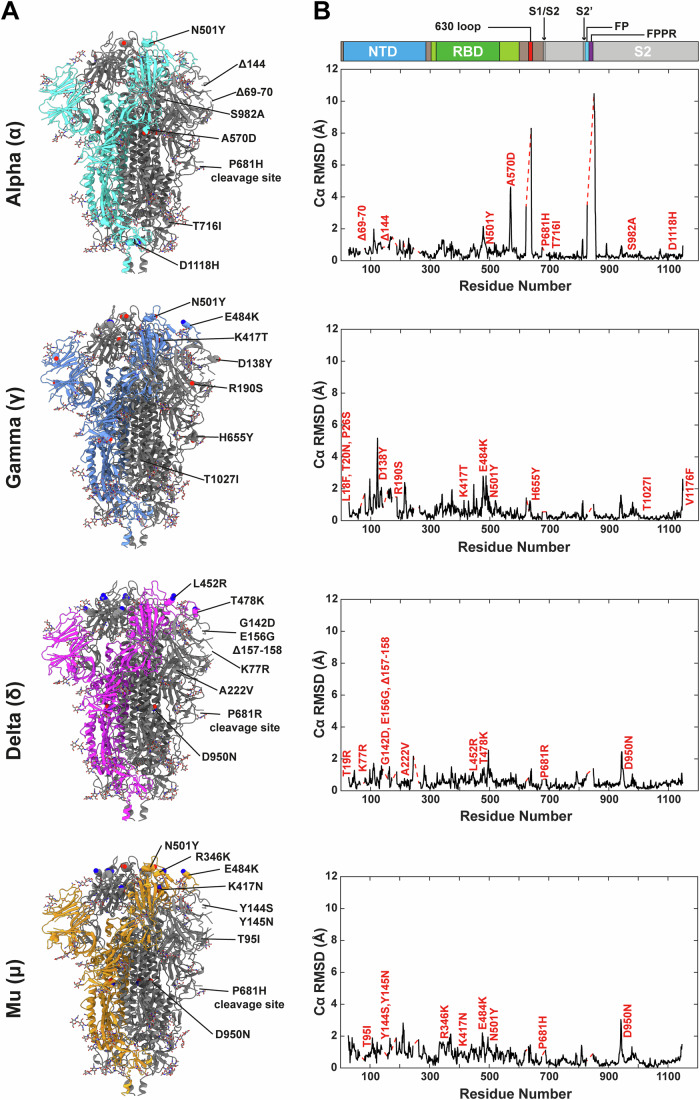
The evolution of SARS-CoV-2 variants with increased fitness has been accompanied by structural changes in the spike (S) proteins, which are the major target for the adaptive immune response. Single-particle cryo-EM analysis of soluble S protein from SARS-CoV-2 variants has revealed this structural adaptation at high resolution. The analysis of S trimers in situ on intact virions has the potential to provide more functionally relevant insights into S structure and virion morphology. Here, we characterized B.1, Alpha, Beta, Gamma, Delta, Kappa, and Mu variants by cryo-electron microscopy and tomography, assessing S cleavage, virion morphology, S incorporation, "in-situ" high-resolution S structures, and the range of S conformational states. We found no evidence for adaptive changes in virion morphology, but describe multiple different positions in the S protein where amino acid changes alter local protein structure. Taken together, our data are consistent with a model where amino acid changes at multiple positions from the top to the base of the spike cause structural changes that can modulate the conformational dynamics of the S protein.
Keywords: Coronavirus; Cryo-electron Tomography; Membrane Fusion Protein; Virus Evolution; Virus Structure.
© 2024. The Author(s).
Conflict of interest statement
Disclosure and competing interests statement. The authors declare no competing interests.
Figures








References
-
- Brown JC, Goldhill DH, Zhou J, Peacock TP, Frise R, Goonawardane N, Baillon L, Kugathasan R, Pinto AL, McKay PF et al (2021) Increased transmission of SARS-CoV-2 lineage B.1.1.7 (VOC 2020212/01) is not accounted for by a replicative advantage in primary airway cells or antibody escape. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/2021.02.24.432576v2 - DOI
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
