

Mechanism of RSL3-induced ferroptotic cell death in HT22 cells: crucial role of protein disulfide isomerase
- PMID: 39544002
- PMCID: PMC12040763
- DOI: 10.3724/abbs.2024165
Mechanism of RSL3-induced ferroptotic cell death in HT22 cells: crucial role of protein disulfide isomerase
Abstract
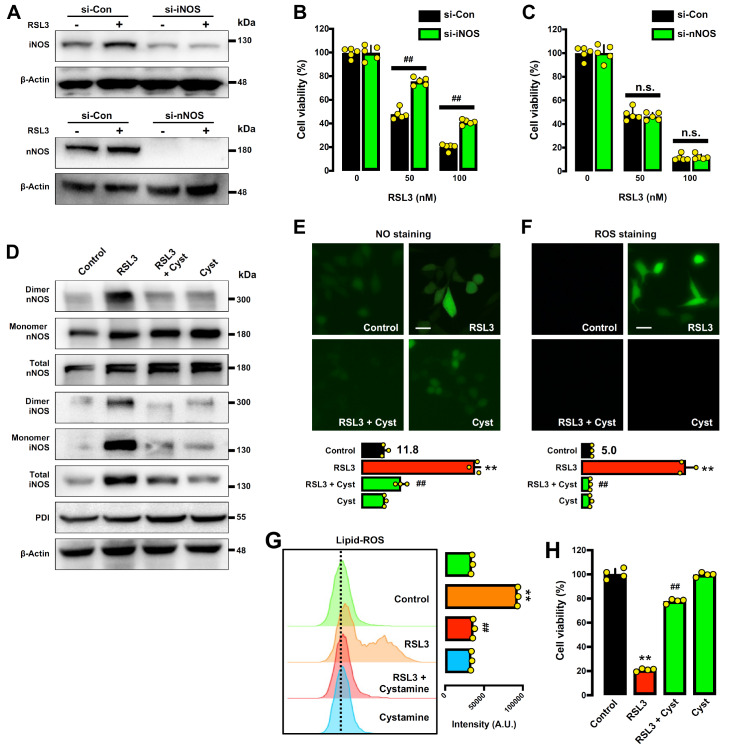
Protein disulfide isomerase (PDI) was recently shown to be an upstream mediator of erastin-induced, glutathione depletion-associated ferroptosis through its catalysis of nitric oxide synthase (NOS) dimerization and nitric oxide (NO) accumulation. A recent study reported that RSL3, a known ferroptosis inducer and glutathione peroxidase 4 (GPX4) inhibitor, can inhibit thioredoxin reductase 1 (TrxR1). The present study seeks to test the hypothesis that RSL3 may, through its inhibition of TrxR1, facilitate PDI activation ( i. e., in a catalytically active, oxidized state), thereby enhancing RSL3-induced ferroptosis through NOS dimerization and NO accumulation. Using HT22 mouse neuronal cells as an in vitro model, we show that treatment of these cells with RSL3 strongly increases NOS protein levels and that PDI-mediated NOS dimerization is activated by RSL3, resulting in NO accumulation. Mechanistically, we find that PDI is activated in cells treated with RSL3 because of its inhibition of TrxR1, and the activated PDI then catalyzes NOS dimerization, which is followed by the accumulation of cellular NO, ROS and lipid-ROS and ultimately ferroptotic cell death. Genetic or pharmacological inhibition of PDI or TrxR1 partially abrogates RSL3-induced NOS activation and the subsequent accumulation of cellular NO, ROS/lipid-ROS, and ultimately ferroptosis in HT22 cells. The results of this study clearly show that PDI activation resulted from RSL3 inhibition of TrxR1 activity contributes crucially to RSL3-induced ferroptosis in a cell culture model through the PDI→NOS→NO→ROS/lipid-ROS pathway, in addition to its known inhibition of GPX4 activity.
Keywords: lipid reactive oxygen species; neuronal nitric oxide synthase; nitric oxide; protein disulfide isomerase; reactive oxygen species; thioredoxin reductase 1.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures








References
-
- Cheff DM, Huang C, Scholzen KC, Gencheva R, Ronzetti MH, Cheng Q, Hall MD, et al. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. . 2023;62:102703. doi: 10.1016/j.redox.2023.102703. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
