

Generating pregnant patient biological profiles by deconvoluting clinical records with electronic health record foundation models
- PMID: 39545787
- PMCID: PMC11565587
- DOI: 10.1093/bib/bbae574
Generating pregnant patient biological profiles by deconvoluting clinical records with electronic health record foundation models
Abstract
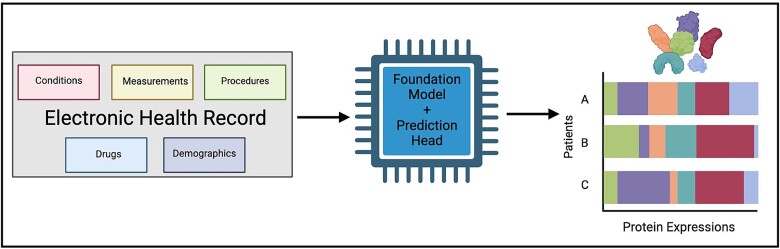
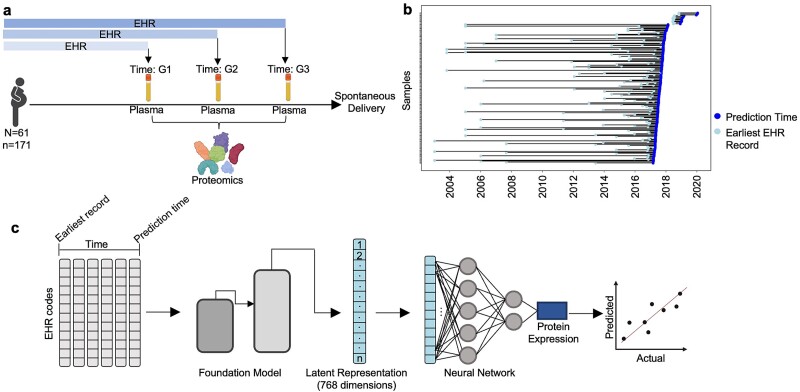
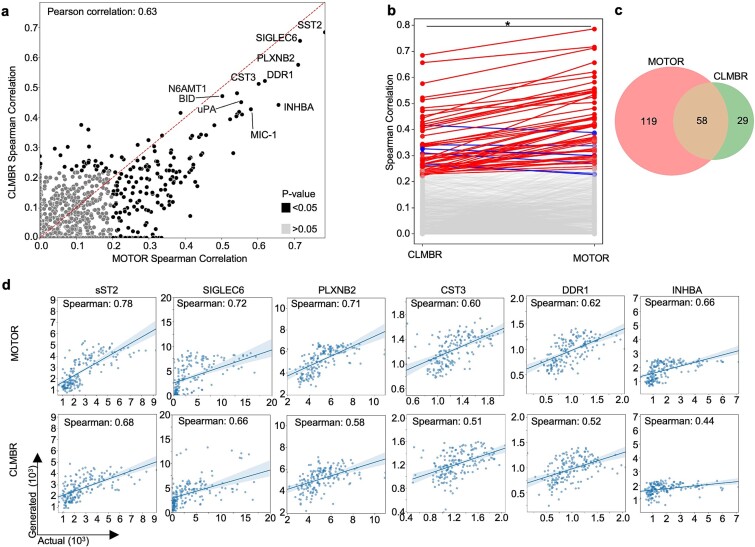
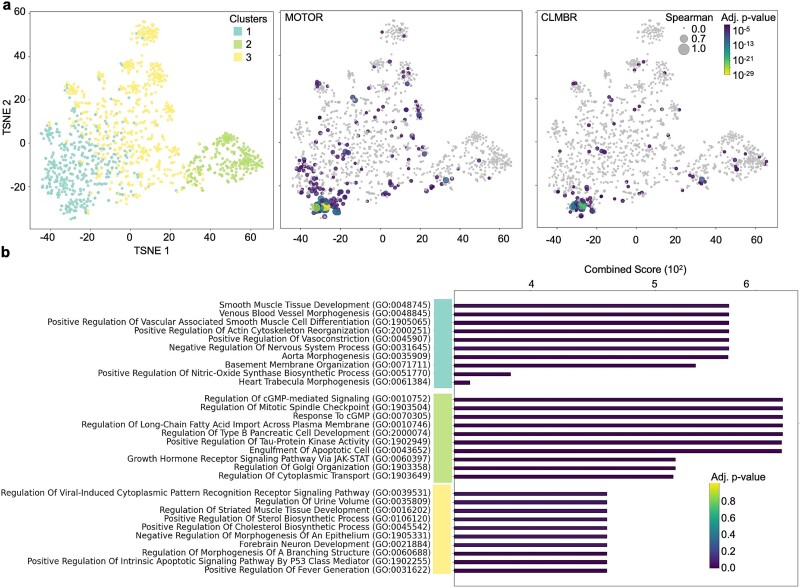
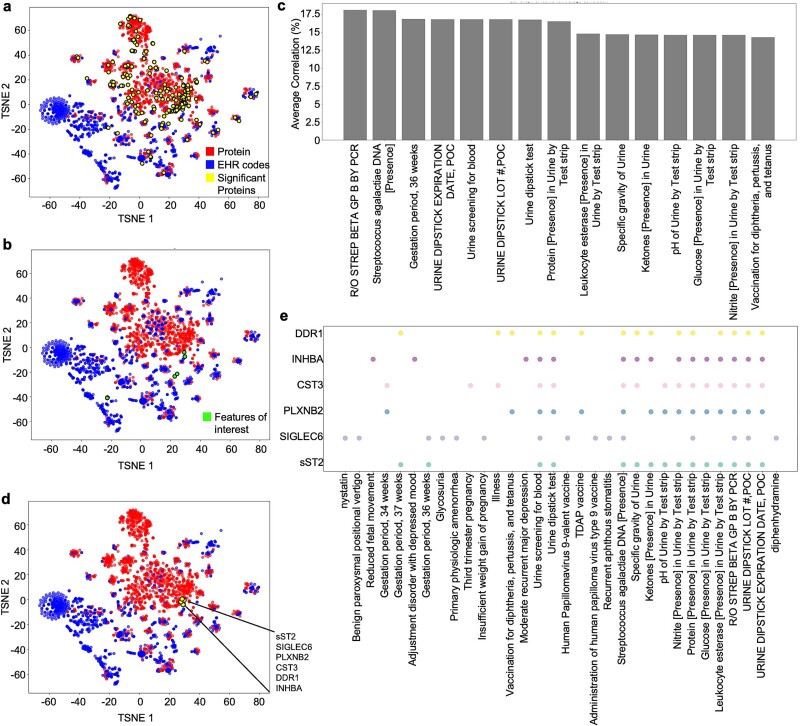
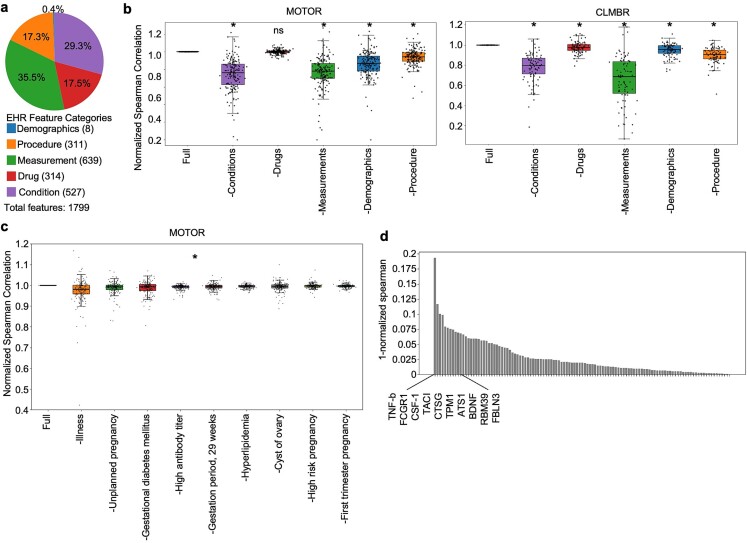
Translational biology posits a strong bi-directional link between clinical phenotypes and a patient's biological profile. By leveraging this bi-directional link, we can efficiently deconvolute pre-existing clinical information into biological profiles. However, traditional computational tools are limited in their ability to resolve this link because of the relatively small sizes of paired clinical-biological datasets for training and the high dimensionality/sparsity of tabular clinical data. Here, we use state-of-the-art foundation models (FMs) for electronic health record (EHR) data to generate proteomics profiles of pregnant patients, thereby deconvoluting pre-existing clinical information into biological profiles without the cost and effort of running large-scale traditional omics studies. We show that FM-derived representations of a patient's EHR data coupled with a fully connected neural network prediction head can generate 206 blood protein expression levels. Interestingly, these proteins were enriched for developmental pathways, while proteins not able to be generated from EHR data were enriched for metabolic pathways. Finally, we show a proteomic signature of gestational diabetes that includes proteins with established and novel links to gestational diabetes. These results showcase the power of FM-derived EHR representations in efficiently generating biological states of pregnant patients. This capability can revolutionize disease understanding and therapeutic development, offering a cost-effective, time-efficient, and less invasive alternative to traditional methods of generating proteomics.
Keywords: electronic health record; foundation model; machine learning; pregnancy; proteomics.
© The Author(s) 2024. Published by Oxford University Press.
Figures
Similar articles
-
Oral anti-diabetic agents for women with established diabetes/impaired glucose tolerance or previous gestational diabetes planning pregnancy, or pregnant women with pre-existing diabetes.Cochrane Database Syst Rev. 2017 Oct 18;10(10):CD007724. doi: 10.1002/14651858.CD007724.pub3. Cochrane Database Syst Rev. 2017. PMID: 29045765 Free PMC article.
-
Deciphering Shared Gene Signatures and Immune Infiltration Characteristics Between Gestational Diabetes Mellitus and Preeclampsia by Integrated Bioinformatics Analysis and Machine Learning.Reprod Sci. 2025 Jun;32(6):1886-1904. doi: 10.1007/s43032-025-01847-1. Epub 2025 May 15. Reprod Sci. 2025. PMID: 40374866
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
Continuous subcutaneous insulin infusion versus multiple daily injections of insulin for pregnant women with diabetes.Cochrane Database Syst Rev. 2016 Jun 7;2016(6):CD005542. doi: 10.1002/14651858.CD005542.pub3. Cochrane Database Syst Rev. 2016. PMID: 27272351 Free PMC article.
Cited by
-
A machine learning approach to leveraging electronic health records for enhanced omics analysis.Nat Mach Intell. 2025;7(2):293-306. doi: 10.1038/s42256-024-00974-9. Epub 2025 Jan 16. Nat Mach Intell. 2025. PMID: 40008295 Free PMC article.
-
AI-guided precision parenteral nutrition for neonatal intensive care units.Nat Med. 2025 Jun;31(6):1882-1894. doi: 10.1038/s41591-025-03601-1. Epub 2025 Mar 25. Nat Med. 2025. PMID: 40133525 Free PMC article.
-
Advancing neonatal health: the promise and challenges of universal genome sequencing in newborn screening.Pediatr Res. 2025 Mar;97(4):1258-1260. doi: 10.1038/s41390-025-03874-9. Epub 2025 Jan 20. Pediatr Res. 2025. PMID: 39833347 No abstract available.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
