

Microbial species and intraspecies units exist and are maintained by ecological cohesiveness coupled to high homologous recombination
- PMID: 39548060
- PMCID: PMC11568254
- DOI: 10.1038/s41467-024-53787-0
Microbial species and intraspecies units exist and are maintained by ecological cohesiveness coupled to high homologous recombination
Abstract
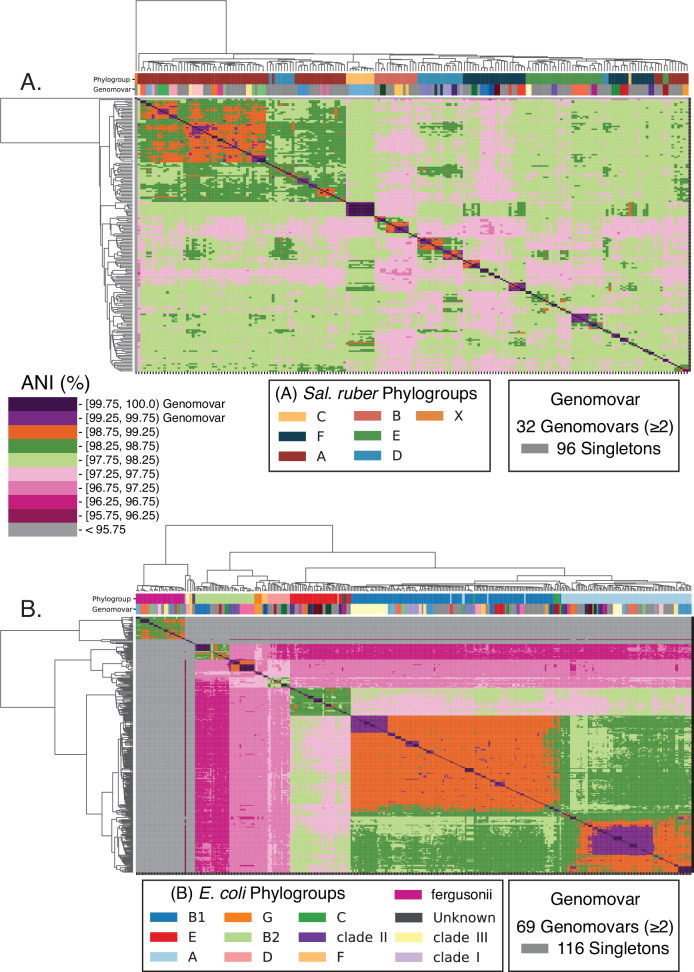
Recent genomic analyses have revealed that microbial communities are predominantly composed of persistent, sequence-discrete species and intraspecies units (genomovars), but the mechanisms that create and maintain these units remain unclear. By analyzing closely-related isolate genomes from the same or related samples and identifying recent recombination events using a novel bioinformatics methodology, we show that high ecological cohesiveness coupled to frequent-enough and unbiased (i.e., not selection-driven) horizontal gene flow, mediated by homologous recombination, often underlie these diversity patterns. Ecological cohesiveness was inferred based on greater similarity in temporal abundance patterns of genomes of the same vs. different units, and recombination was shown to affect all sizable segments of the genome (i.e., be genome-wide) and have two times or greater impact on sequence evolution than point mutations. These results were observed in both Salinibacter ruber, an environmental halophilic organism, and Escherichia coli, the model gut-associated organism and an opportunistic pathogen, indicating that they may be more broadly applicable to the microbial world. Therefore, our results represent a departure compared to previous models of microbial speciation that invoke either ecology or recombination, but not necessarily their synergistic effect, and answer an important question for microbiology: what a species and a subspecies are.
© 2024. The Author(s).
Conflict of interest statement
Figures




References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
