

Subcellular mRNA kinetic modeling reveals nuclear retention as rate-limiting
- PMID: 39548324
- PMCID: PMC11611909
- DOI: 10.1038/s44320-024-00073-2
Subcellular mRNA kinetic modeling reveals nuclear retention as rate-limiting
Abstract
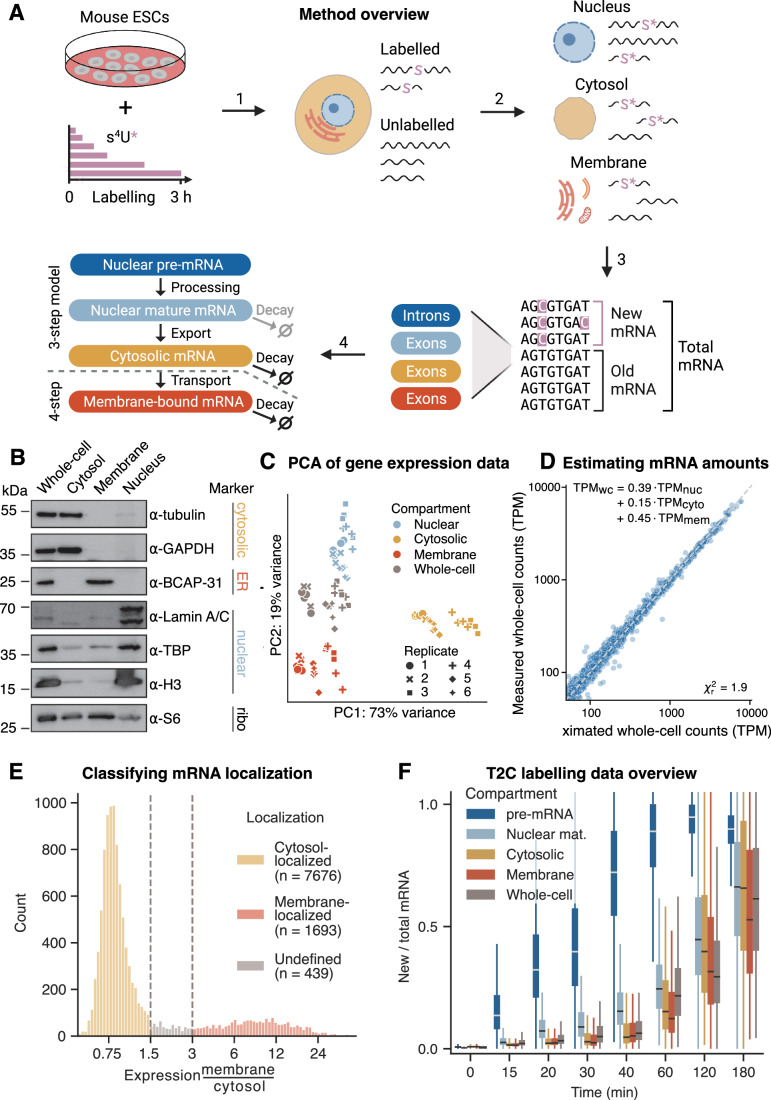
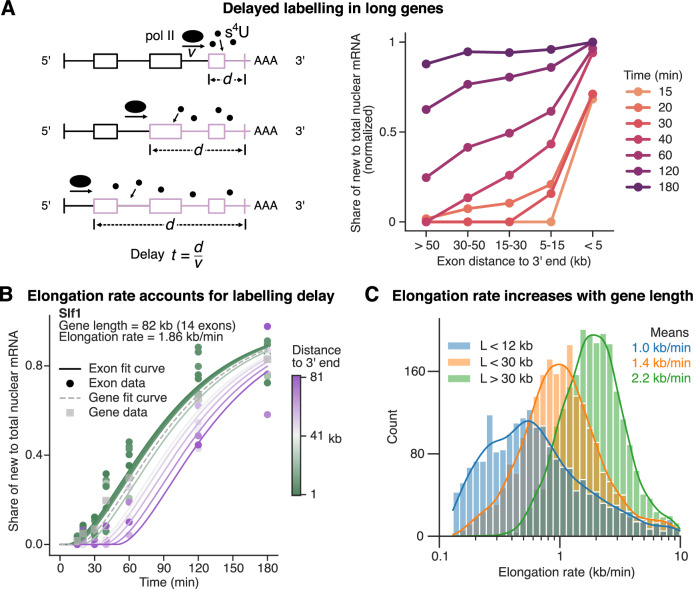
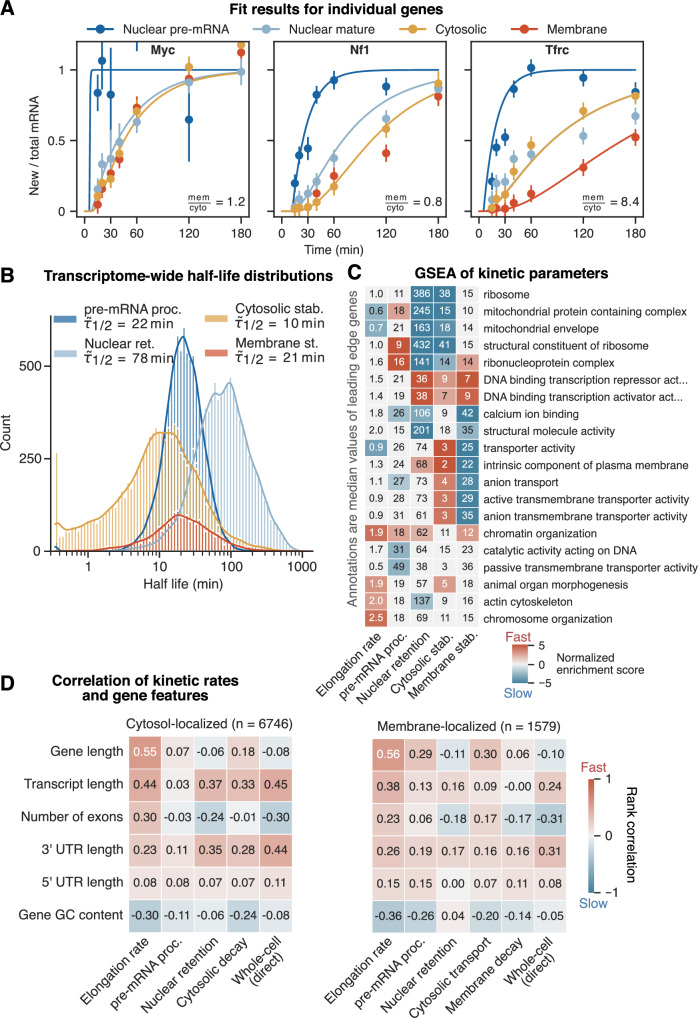
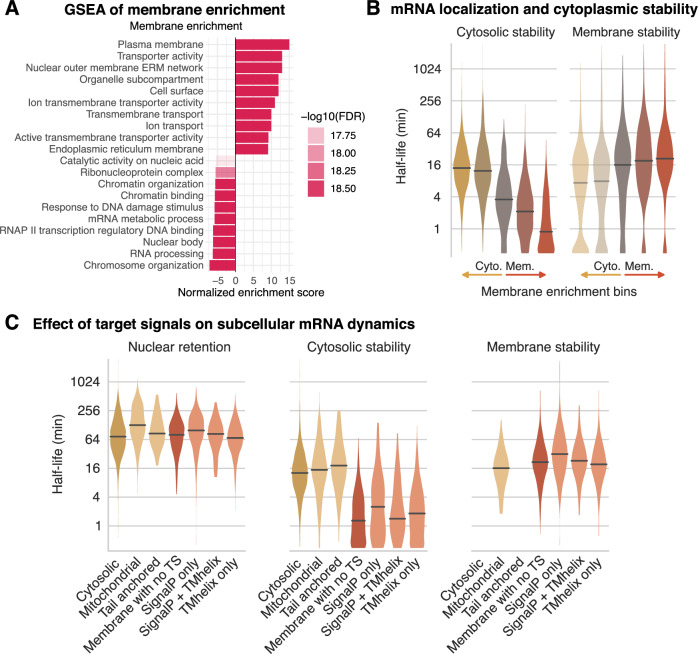
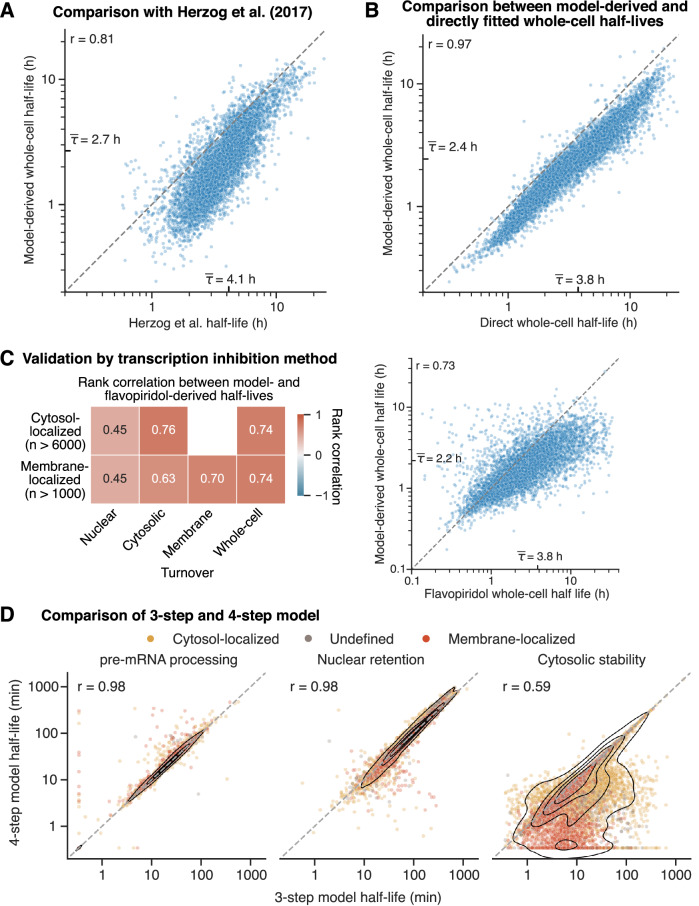
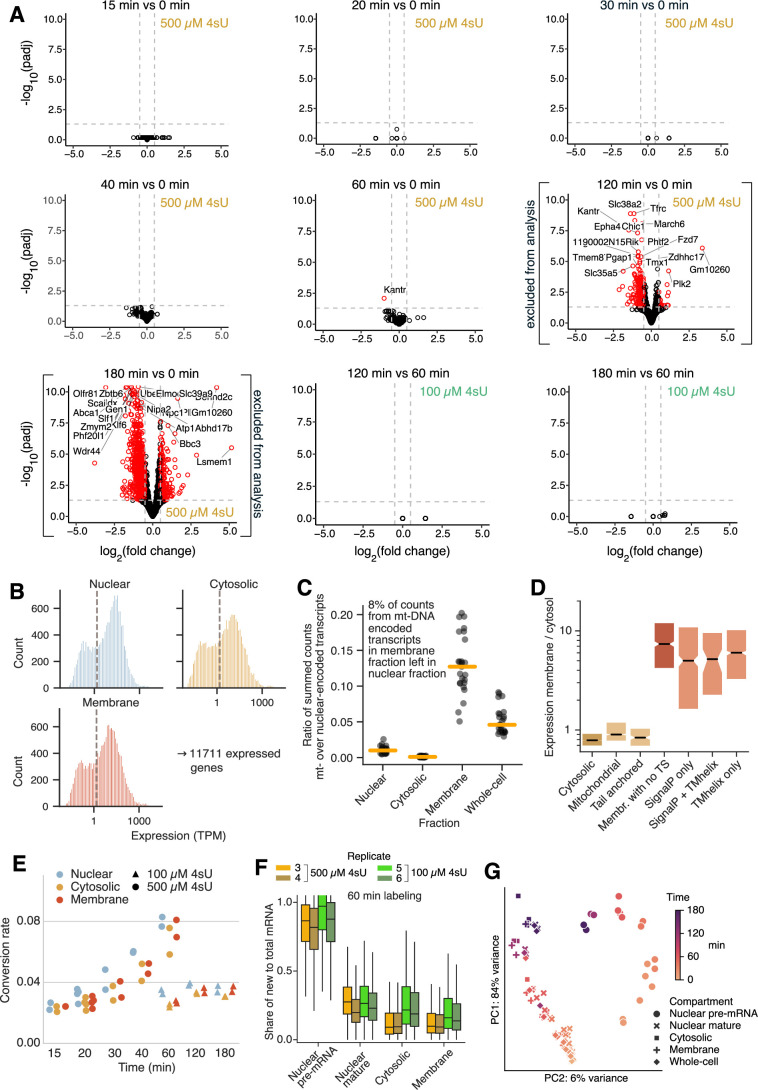
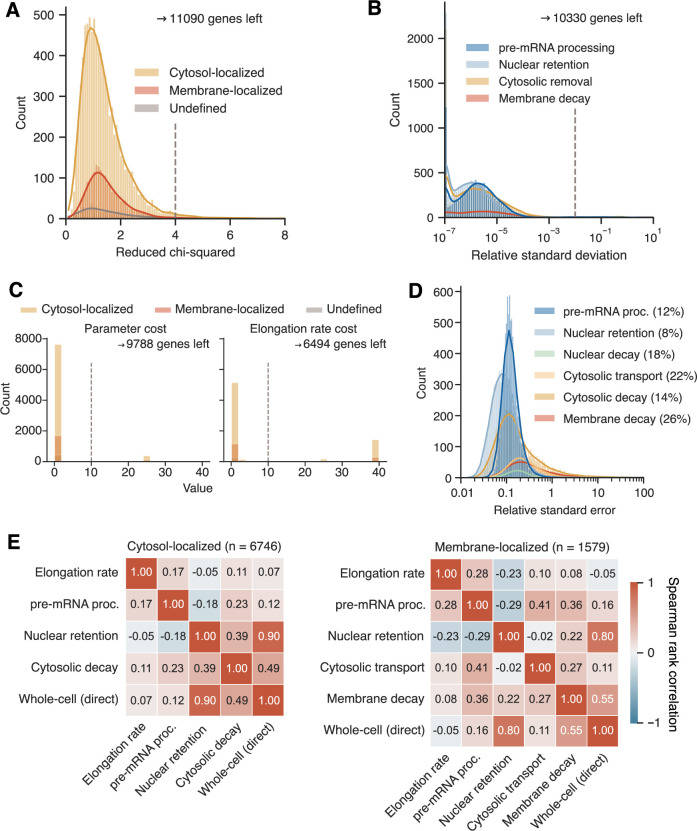
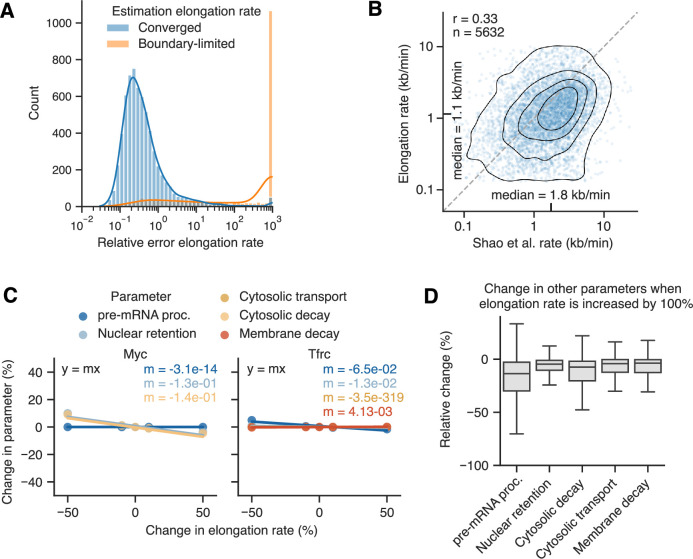
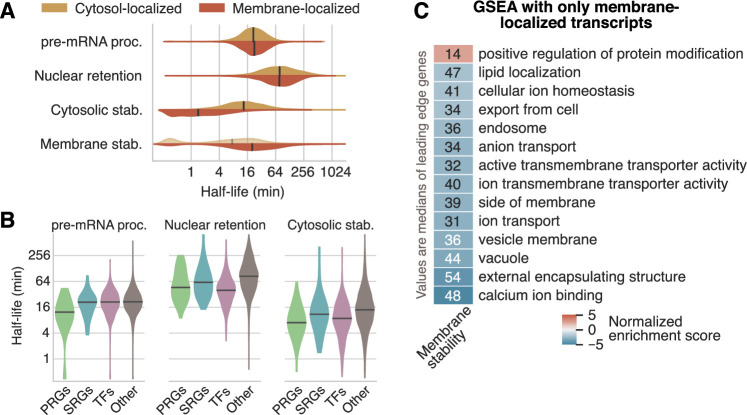
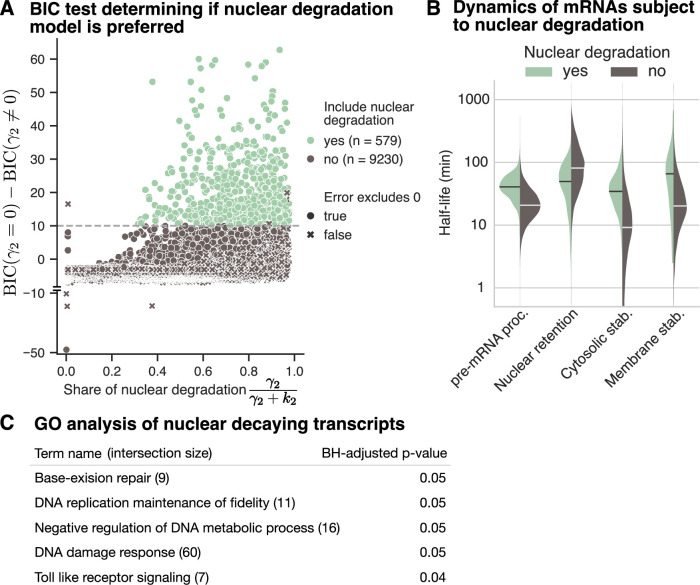
Eukaryotic mRNAs are transcribed, processed, translated, and degraded in different subcellular compartments. Here, we measured mRNA flow rates between subcellular compartments in mouse embryonic stem cells. By combining metabolic RNA labeling, biochemical fractionation, mRNA sequencing, and mathematical modeling, we determined the half-lives of nuclear pre-, nuclear mature, cytosolic, and membrane-associated mRNAs from over 9000 genes. In addition, we estimated transcript elongation rates. Many matured mRNAs have long nuclear half-lives, indicating nuclear retention as the rate-limiting step in the flow of mRNAs. In contrast, mRNA transcripts coding for transcription factors show fast kinetic rates, and in particular short nuclear half-lives. Differentially localized mRNAs have distinct rate constant combinations, implying modular regulation. Membrane stability is high for membrane-localized mRNA and cytosolic stability is high for cytosol-localized mRNA. mRNAs encoding target signals for membranes have low cytosolic and high membrane half-lives with minor differences between signals. Transcripts of nuclear-encoded mitochondrial proteins have long nuclear retention and cytoplasmic kinetics that do not reflect co-translational targeting. Our data and analyses provide a useful resource to study spatiotemporal gene expression regulation.
Keywords: Kinetic Modeling; Metabolic Labeling; Nuclear Retention; RNA Dynamics; Subcellular Fractionation.
© 2024. The Author(s).
Conflict of interest statement
Disclosure and competing interests statement. The authors declare no competing interests.
Figures
Similar articles
-
Nuclear export is a limiting factor in eukaryotic mRNA metabolism.PLoS Comput Biol. 2024 May 16;20(5):e1012059. doi: 10.1371/journal.pcbi.1012059. eCollection 2024 May. PLoS Comput Biol. 2024. PMID: 38753883 Free PMC article.
-
Comprehensive analysis of the circadian nuclear and cytoplasmic transcriptome in mouse liver.PLoS Genet. 2022 Aug 3;18(8):e1009903. doi: 10.1371/journal.pgen.1009903. eCollection 2022 Aug. PLoS Genet. 2022. PMID: 35921362 Free PMC article.
-
Characterization of the nuclear and cytosolic transcriptomes in human brain tissue reveals new insights into the subcellular distribution of RNA transcripts.Sci Rep. 2021 Feb 18;11(1):4076. doi: 10.1038/s41598-021-83541-1. Sci Rep. 2021. PMID: 33603054 Free PMC article.
-
Nuclear retention of mRNAs - quality control, gene regulation and human disease.Semin Cell Dev Biol. 2018 Jul;79:131-142. doi: 10.1016/j.semcdb.2017.11.001. Epub 2017 Nov 22. Semin Cell Dev Biol. 2018. PMID: 29102717 Review.
-
Localization of RNAs in the nucleus: cis- and trans- regulation.RNA Biol. 2021 Dec;18(12):2073-2086. doi: 10.1080/15476286.2021.1894025. Epub 2021 Mar 8. RNA Biol. 2021. PMID: 33682620 Free PMC article. Review.
Cited by
-
Halfpipe: a tool for analyzing metabolic labeling RNA-seq data to quantify RNA half-lives.NAR Genom Bioinform. 2025 Feb 18;7(1):lqaf006. doi: 10.1093/nargab/lqaf006. eCollection 2025 Mar. NAR Genom Bioinform. 2025. PMID: 39967604 Free PMC article.
-
Expanding and improving analyses of nucleotide recoding RNA-seq experiments with the EZbakR suite.PLoS Comput Biol. 2025 Jul 3;21(7):e1013179. doi: 10.1371/journal.pcbi.1013179. eCollection 2025 Jul. PLoS Comput Biol. 2025. PMID: 40609070 Free PMC article.
-
Expanding and improving analyses of nucleotide recoding RNA-seq experiments with the EZbakR suite.bioRxiv [Preprint]. 2024 Oct 17:2024.10.14.617411. doi: 10.1101/2024.10.14.617411. bioRxiv. 2024. Update in: PLoS Comput Biol. 2025 Jul 3;21(7):e1013179. doi: 10.1371/journal.pcbi.1013179. PMID: 39463977 Free PMC article. Updated. Preprint.
-
Gene-specific transcript buffering revealed by perturbation of coactivator complexes.Sci Adv. 2025 Mar 21;11(12):eadr1492. doi: 10.1126/sciadv.adr1492. Epub 2025 Mar 19. Sci Adv. 2025. PMID: 40106549 Free PMC article.
References
-
- Battich N, Stoeger T, Pelkmans L (2015) Control of transcript variability in single mammalian cells. Cell 163:1596–1610 - PubMed
-
- Berry S, Pelkmans L (2022) Mechanisms of cellular mRNA transcript homeostasis. Trends Cell Biol 32:655–668 - PubMed
-
- Chao SH, Price DH (2001) Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem 276:31793–31799 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
