

Investigating the diagnostic and prognostic significance of genes related to fatty acid metabolism in hepatocellular carcinoma
- PMID: 39548390
- PMCID: PMC11566841
- DOI: 10.1186/s12876-024-03495-2
Investigating the diagnostic and prognostic significance of genes related to fatty acid metabolism in hepatocellular carcinoma
Abstract
Background: Hepatocellular carcinoma (HCC) is one of the most prevalent and lethal cancers worldwide, with death rates increasing by approximately 2-3% per year. The high mortality and poor prognosis of HCC are primarily due to inaccurate early diagnosis and lack of monitoring when liver transplantation is not feasible. Fatty acid (FA) metabolism is a critical metabolic pathway that provides energy and signaling factors in cancer, particularly in HCC, and promotes malignancy. Therefore, it is essential to explore specific FA metabolism-related diagnostic and prognostic signatures that can enable the effective early diagnosis and monitoring of HCC.
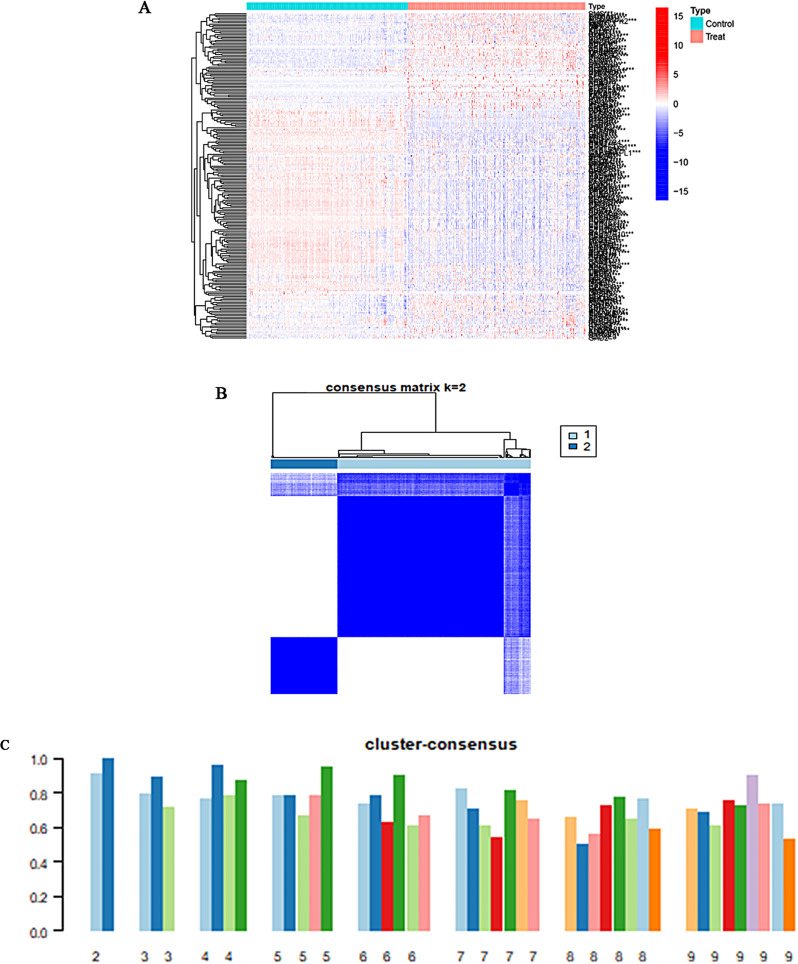
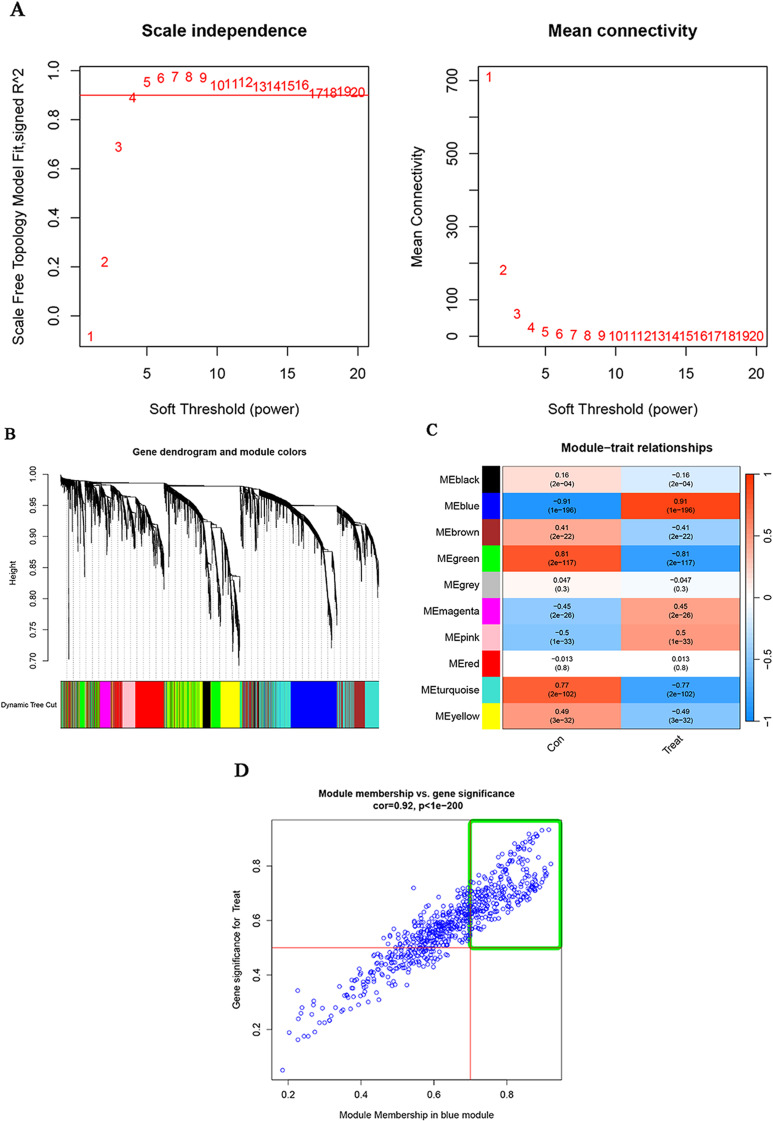
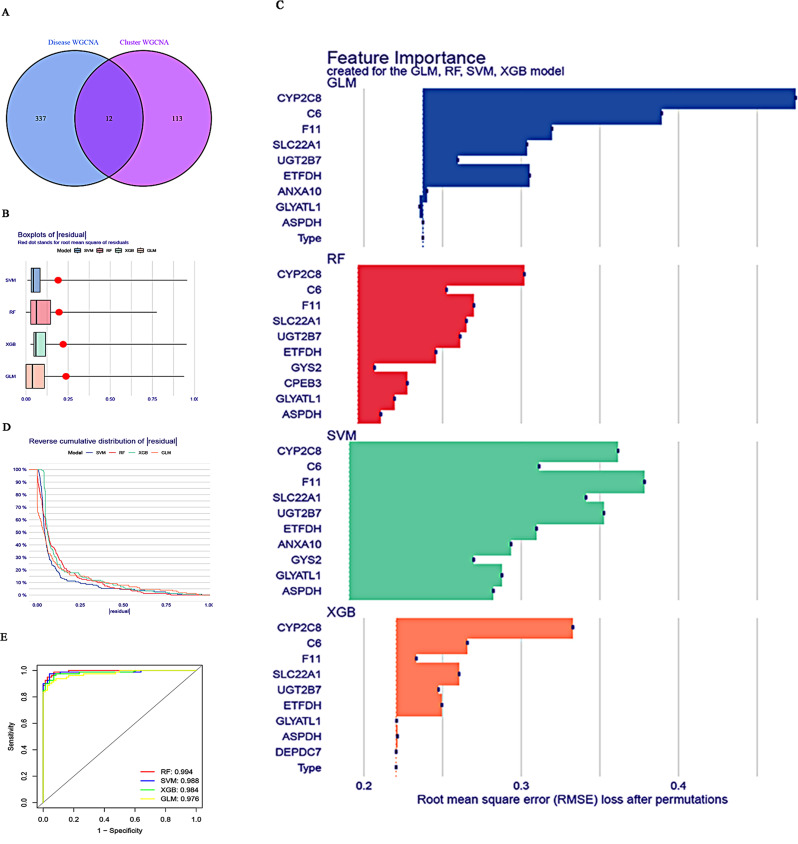
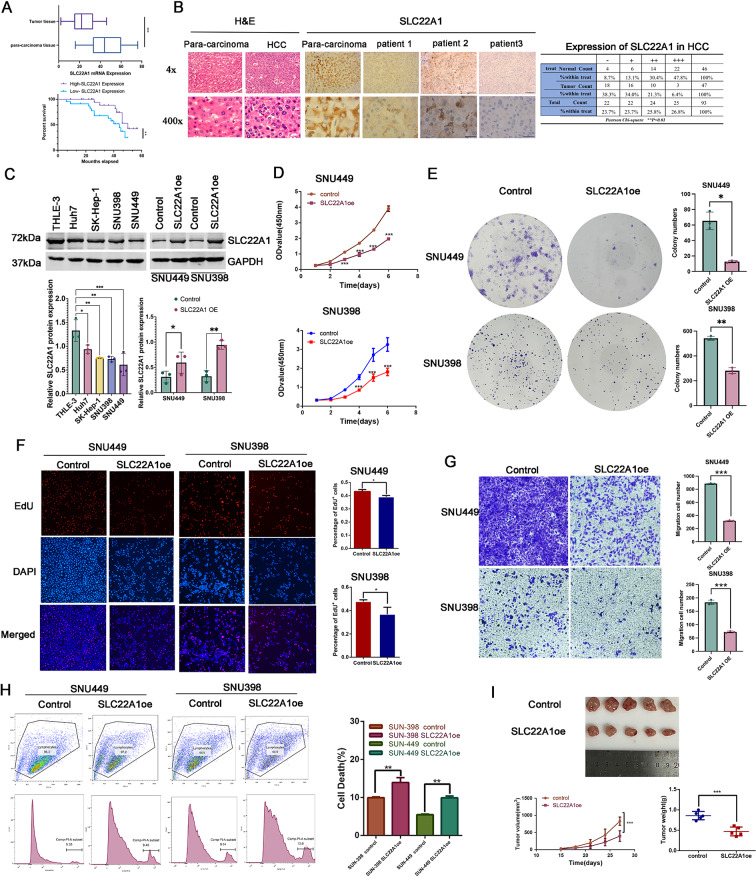
Methods: In this study, we used genes associated with FA metabolism pathway and weighted gene co-expression network analysis (WGCNA) to establish a gene co-expression network and identify hub genes related to HCC (disease WGCNA) and FA clusters (cluster WGCNA). A diagnostic model was constructed using data downloaded from the Gene Expression Omnibus database (GSE25097), and a prognostic model was established using The Cancer Genome Atlas cohort, in which Univariate Cox regression analysis, multivariate Cox risk model, and LASSO Cox regression analysis were applied. The immune infiltration of HCC cells was evaluated using CIBERSORT. The function of the key SLC22A1 gene was experimentally verified in vitro and in vivo.
Results: Twelve overlapping genes (CPEB3, ASPDH, DEPDC7, ETFDH, UGT2B7, GYS2, F11, ANXA10, CYP2C8, GLYATL1, C6, and SLC22A1) from disease and cluster WGCNA were identified as key genes and used in the construction of the diagnostic and prognostic models. The RF model had the highest area under the ROC curve (AUC) of 0.994 was identified as the most effective for distinguishing patients with HCC with different features. The top five important genes (C6, UGT2B7, SLC22A1, F11, and CYP2C8) from the RF model were selected as diagnostic genes for further analysis (ROC curves: AUC value = 0.986, 95% confidence interval [95% CI] = 0.967-0.999). Moreover, a risk score formula consisting of four genes (GYS2, F11, ANXA10 and SLC22A1) was established and its independent prognostic ability was further demonstrated (univariate Cox regression analysis: hazard ratio [HR] = 3.664%, 95% CI = 2.033-6.605, P < 0.001; multivariate Cox regression analysis: HR = 2.801%, 95% CI = 1.553-5.049, P < 0.001). Additionally, in vitro and in vivo experiments demonstrated that SLC22A1 inhibits HCC tumor development, suggesting it may be a potential therapeutic target for HCC.
Conclusions: These findings indicate a considerable value of specific FA metabolism-related genes in the diagnostic and prognostic evaluation of HCC, which provide novel insights into the disease's management, as well as has potential implications for personalized treatment strategies. However, further investigation of the effects of these model genes on HCC is required.
Keywords: SLC22A1; Biomarker; Diagnosis; Fatty acid metabolism-related genes; Hepatocellular carcinoma; Prognosis; Weighted gene co-expression network analysis.
© 2024. The Author(s).
Conflict of interest statement
Figures






References
-
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. Cancer J Clin. 2021;71(3):209–49. - PubMed
-
- Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–63. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
