

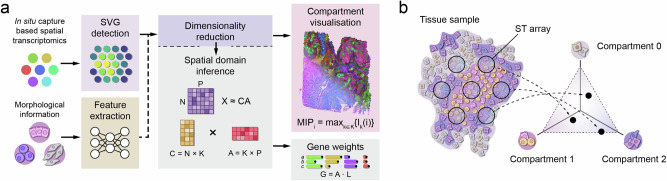
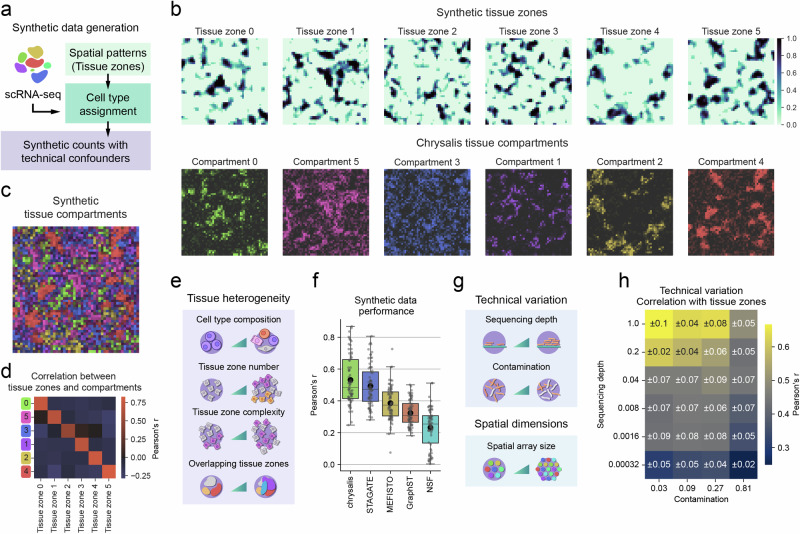
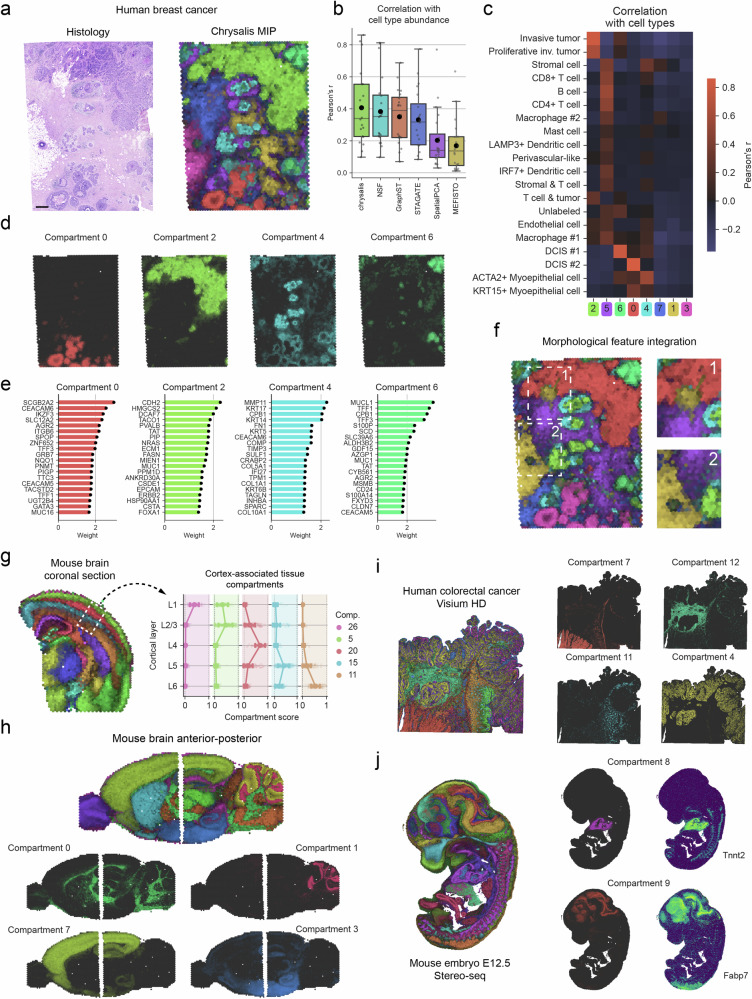
Chrysalis: decoding tissue compartments in spatial transcriptomics with archetypal analysis
- PMID: 39550461
- PMCID: PMC11569261
- DOI: 10.1038/s42003-024-07165-7
Chrysalis: decoding tissue compartments in spatial transcriptomics with archetypal analysis
Abstract
Dissecting tissue compartments in spatial transcriptomics (ST) remains challenging due to limited spatial resolution and dependence on single-cell reference data. We present Chrysalis, a computational method that rapidly uncovers tissue compartments through spatially variable gene (SVG) detection and archetypal analysis without requiring external reference data. Additionally, it offers a unique visualisation approach for swift tissue characterisation and provides access to the underlying gene expression signatures, enabling the identification of spatially and functionally distinct cellular niches. Chrysalis was evaluated through various benchmarks and validated against deconvolution, independently obtained cell type abundance data, and histopathological annotations, demonstrating superior performance compared to other algorithms on both in silico and real-world test examples. Furthermore, we showcased its versatility across different technologies, such as Visium, Visium HD, Slide-seq, and Stereo-seq.
© 2024. The Author(s).
Conflict of interest statement
Figures

References
-
- Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science353, 78–82 (2016). - PubMed
-
- Cao, J. et al. Decoder-seq enhances mRNA capture efficiency in spatial RNA sequencing. Nat. Biotechnol. 1–12 10.1038/s41587-023-02086-y (2024). - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
