

Expanding the genotypic and phenotypic spectrum of Egyptian children with maple syrup urine disease
- PMID: 39551846
- PMCID: PMC11570613
- DOI: 10.1038/s41598-024-78105-y
Expanding the genotypic and phenotypic spectrum of Egyptian children with maple syrup urine disease
Abstract
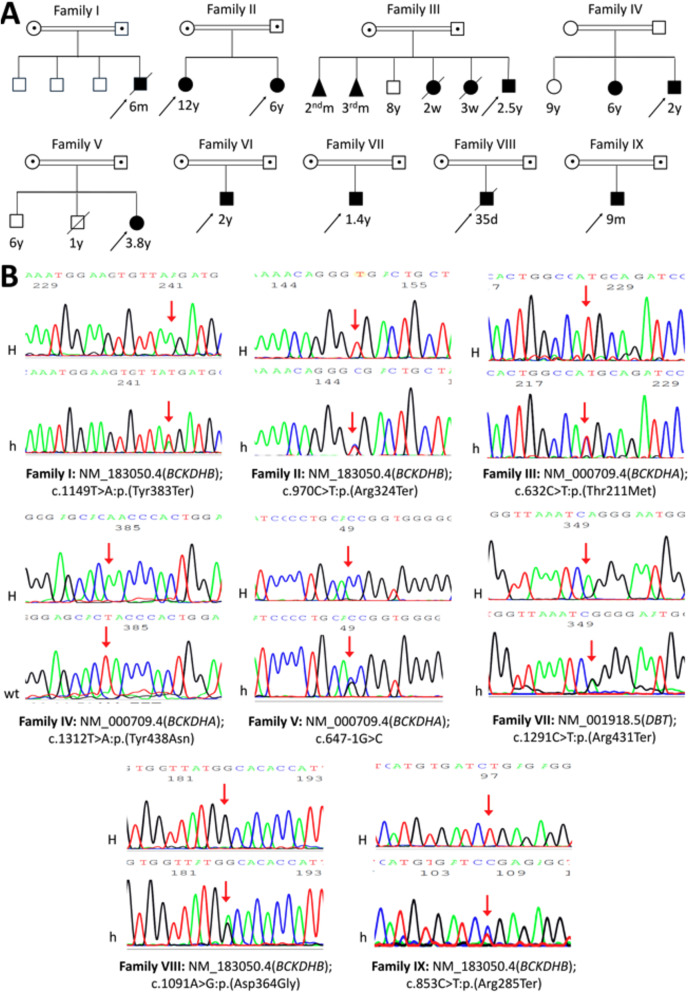
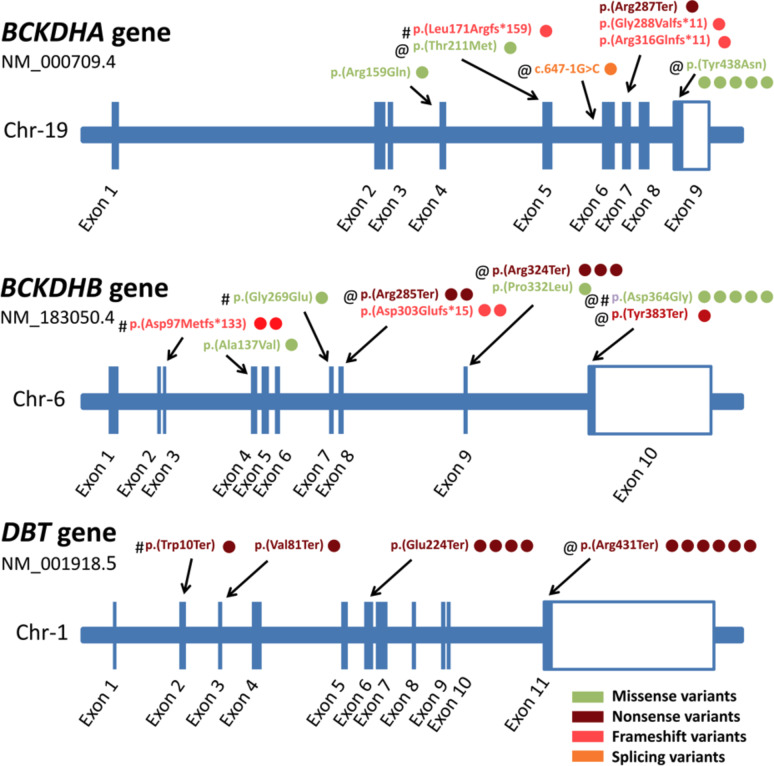
Maple Syrup Urine Disease (MSUD, OMIM# 248600) is an autosomal recessive inborn error of metabolism characterized by elevated branched chain amino acids (BCAA) leucine/isoleucine and valine in blood of affected children. The phenotypic and genotypic spectrum of MSUD is largely unreported in Egypt. We recruited ten patients (4 males/6 females, 2weeks-12years) from nine unrelated families with clinical and biochemical evidence of MSUD. We performed Sanger sequencing for the three most-commonly responsible genes: BCKDHA, BCKDHB and DBT and conducted exome sequencing for unresolved cases. Through Sanger sequencing, we detected eight homozygous pathogenic/likely pathogenic variants (four in BCKDHB, three in BCKDHA and one in DBT gene) in eight different families. The proband of family VI, who had no significant genetic findings by Sanger, had a peculiar phenotype and atypical radiological findings. His exome sequencing revealed a previously reported homozygous likely pathogenic variant in the RARS2 gene (NM_020320.5:c.1026G > A;p.(Met342Ile)) causing the mitochondrial-encephalopathy disorder pontocerebellar hypoplasia, type 6 (OMIM# 611523). Furthermore, the copy-number-variant analysis of the exome data revealed a biallelic duplication affecting exons 2-6 of the BCKDHB gene (GRCh38: Chr.6-g.80127496:80171441dup) evaluated as variant of uncertain significance but expected to cause a breakpoint and may disrupt gene function, which can explain the markedly elevated BCAA levels in the patient's blood. In conclusion, we expanded the genotypic and phenotypic spectrum of the disease and showed that aggressive intervention with specific treatment in the first few days of life resulted in normal development even in a developing country setting. Inclusion of MSUD in the national newborn screening program in Egypt is highly recommended.
Keywords: Egypt; Exome sequencing; Maple-syrup-urine disease; Sanger sequencing; Novel variant.
© 2024. The Author(s).
Conflict of interest statement
Figures
References
-
- Chuang, D. T., Shih, V. E. & Max Wynn, R. R. Maple syrup urine disease (branched-chain ketoaciduria). Valle D.L., &Antonarakis S, & Ballabio A, & Beaudet A.L., & Mitchell G.A.(Eds.), The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill Education. :1971–2006. (2001). https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225084607 (2019).
-
- Jaradat, S. A. et al. Al Rababah, B. Molecular analysis of maple syrup urine disease in Jordanian families. Meta Gene10, 81–85. 10.1016/j.mgene.2015.10.002 (2015). - DOI
-
- Strauss, K. A., Carson, V. J., Soltys, K., Young, M. E., Bowser, L. E., Puffenberger,E. G., Brigatti, K. W., Williams, K. B., Robinson, D. L., Hendrickson, C., Beiler,K., Taylor, C. M., Haas-Givler, B., Chopko, S., Hailey, J., Muelly, E. R., Shellmer,D. A., Radcliff, Z., Rodrigues, A., Loeven, K., … Morton, D. H. Branched-chain α-ketoacid dehydrogenase deficiency (maple syrup urine disease): Treatment, biomarkers,and outcomes. Mol Genet Metab, 129(3), 193–206. 10.1016/j.ymgme.2020.01.006 (2020). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
