

An AMP-activated protein kinase-PGC-1α axis mediates metabolic plasticity in glioblastoma
- PMID: 39552019
- PMCID: PMC11570551
- DOI: 10.1002/ctm2.70030
An AMP-activated protein kinase-PGC-1α axis mediates metabolic plasticity in glioblastoma
Abstract
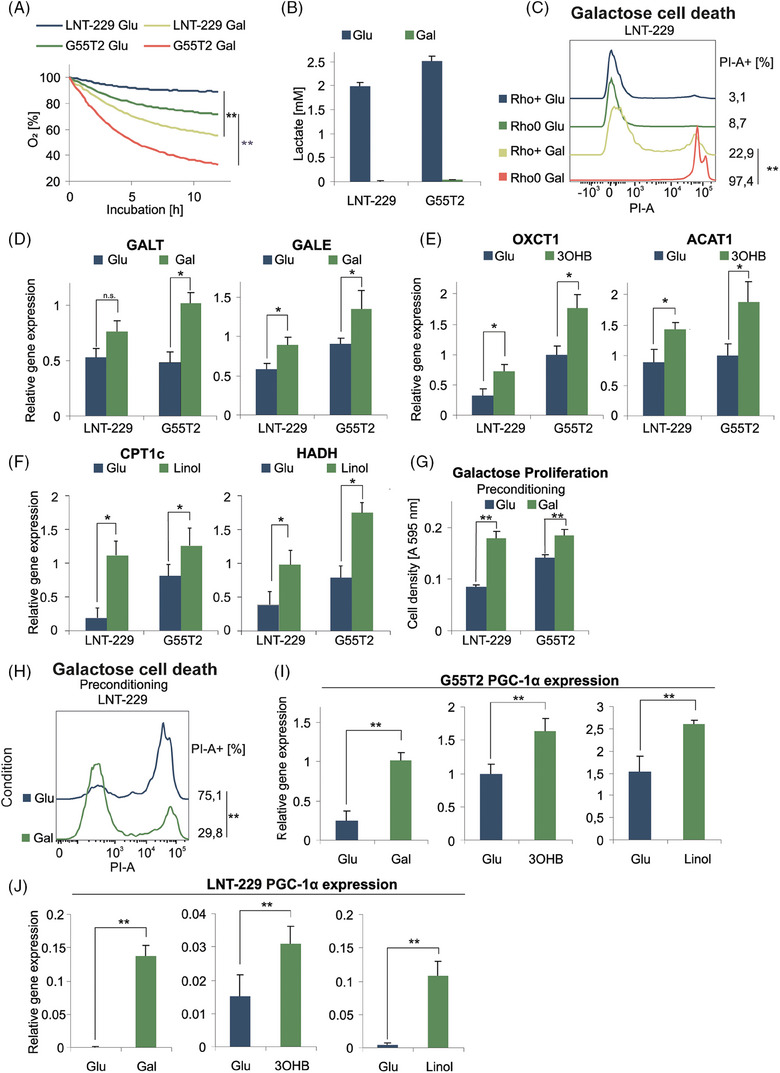
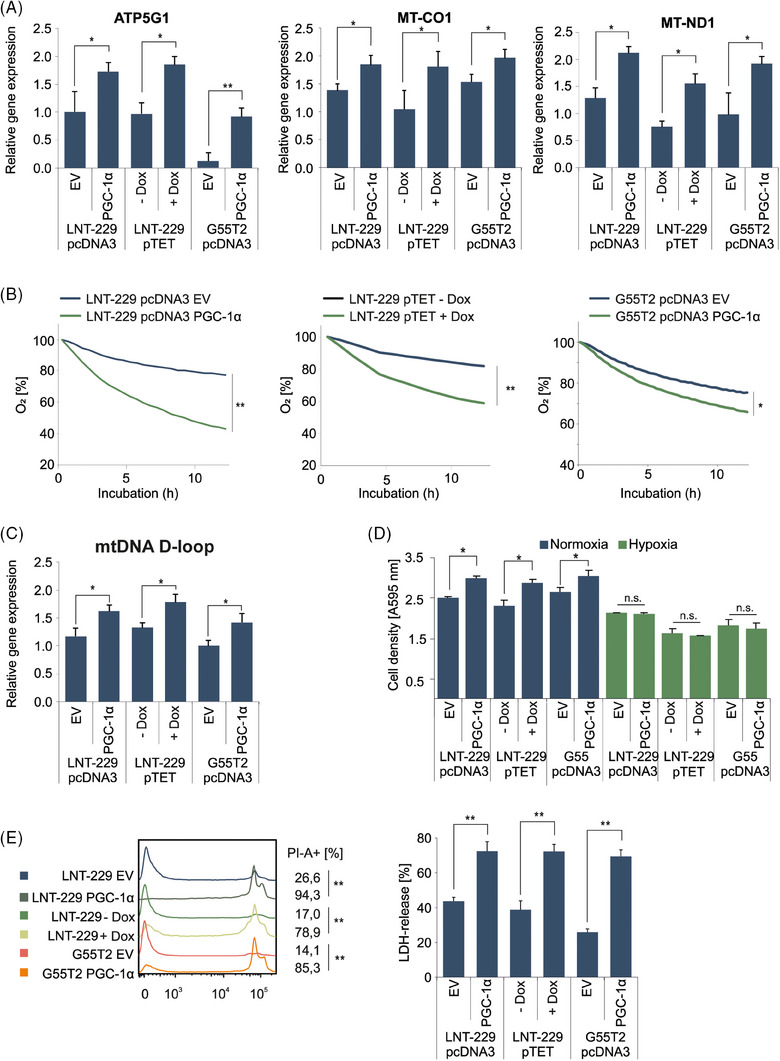
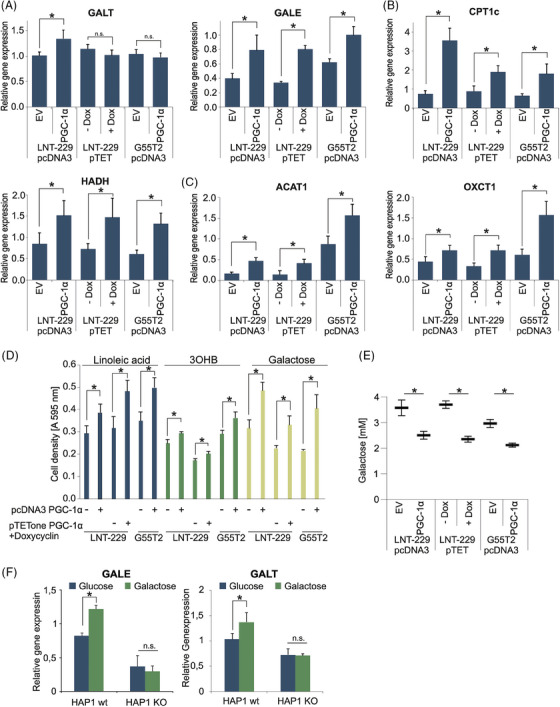
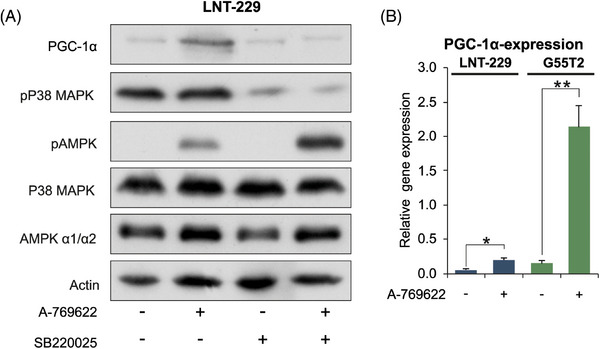
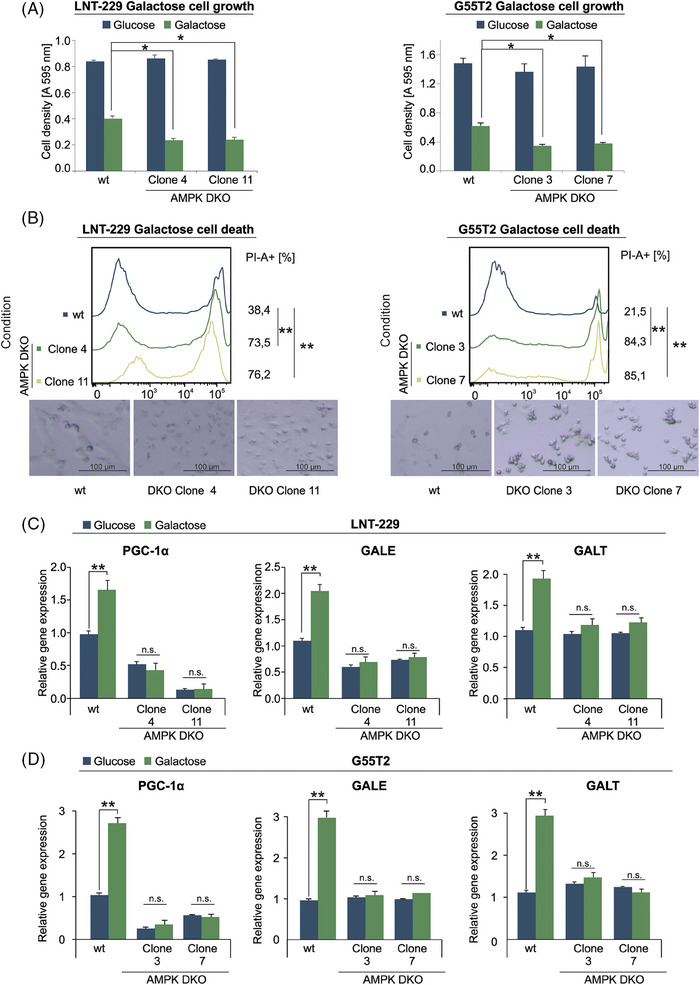
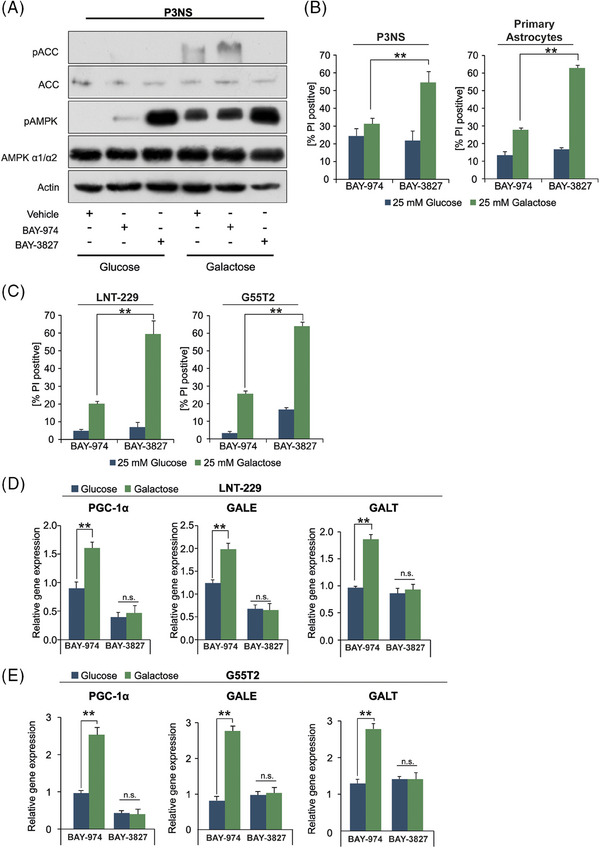
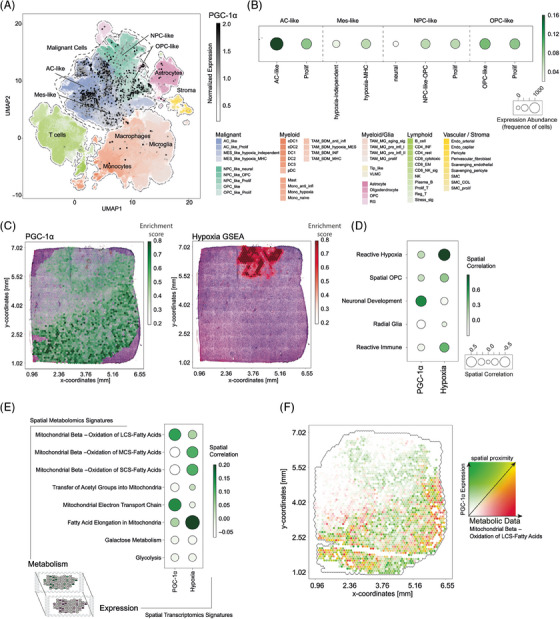
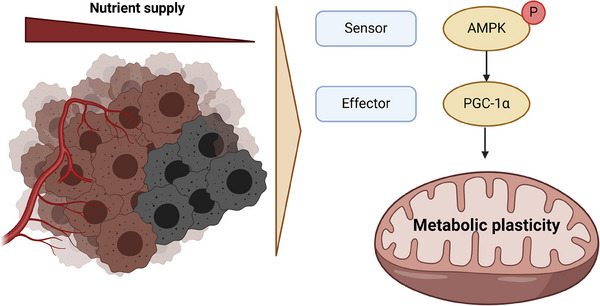
Glioblastoma, the most frequent primary malignant brain tumour in adults, is characterised by profound yet dynamic hypoxia and nutrient depletion. To sustain survival and proliferation, tumour cells are compelled to acquire metabolic plasticity with the induction of adaptive metabolic programs. Here, we interrogated the pathways necessary to enable processing of nutrients other than glucose. We employed genetic approaches (stable/inducible overexpression, CRISPR/Cas9 knockout), pharmacological interventions with a novel inhibitor of AMP-activated protein kinase (AMPK) in glioblastoma cell culture systems and a proteomic approach to investigate mechanisms of metabolic plasticity. Moreover, a spatially resolved multiomic analysis was employed to correlate the gene expression pattern of PGC-1α with the local metabolic and genetic architecture in human glioblastoma tissue sections. A switch from glucose to alternative nutrients triggered an activation of AMPK, which in turn activated PGC-1α-dependent adaptive programs promoting mitochondrial metabolism. This sensor-effector mechanism was essential for metabolic plasticity with both functional AMPK and PGC-1α necessary for survival and growth of cells under nonglucose nutrient sources. In human glioblastoma tissue specimens, PGC-1α-expression correlated with nonhypoxic tumour niches defining a specific metabolic compartment. Our findings reveal a cell-intrinsic nutrient sensing and switching mechanism. The exposure to alternative fuels triggers a starvation signal that subsequently is passed on via AMPK and PGC-1α to induce adaptive programs necessary for broader spectrum nutrient metabolism. The integration of spatially resolved transcriptomic data confirms the relevance of PGC-1α especially in nonhypoxic tumour regions. Thus, the AMPK-PGC-1α axis is a candidate for therapeutic inhibition in glioblastoma. KEY POINTS/HIGHLIGHTS: AMPK activation induces PGC-1α expression in glioblastoma during nutrient scarcity. PGC-1α enables metabolic plasticity by facilitating metabolism of alternative nutrients in glioblastoma. PGC-1α expression is inversely correlated with hypoxic tumour regions in human glioblastomas.
Keywords: AMP‐activated protein kinase; PGC‐1α; PPARGC1A; glioblastoma; hypoxia; metabolic plasticity; tumour microenvironment.
© 2024 The Author(s). Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
JPS reports honoraria for lectures or advisory board participation or consulting or travel grants from Abbvie, Roche, Boehringer, Bristol‐Myers Squibb, Medac, Mundipharma and UCB. Michael W. Ronellenfitsch reports a research grant from UCB as well as honoraria for advisory board participation from Alexion. All other authors declare no conflicts of interest.
Figures
References
-
- Warburg O, Posener K, Negelein E. Über den Stoffwechsel der Carcinomzelle. Biochem Z. 1924(152): 319‐344.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases