

This is a preprint.
WAVE1 and WAVE2 facilitate human papillomavirus-driven actin polymerization during cellular entry
- PMID: 39553927
- PMCID: PMC11565777
- DOI: 10.1101/2024.10.28.620484
WAVE1 and WAVE2 facilitate human papillomavirus-driven actin polymerization during cellular entry
Update in
-
Human Papillomavirus Type 16 Stimulates WAVE1- and WAVE2-Dependent Actin Protrusions for Endocytic Entry.Viruses. 2025 Apr 8;17(4):542. doi: 10.3390/v17040542. Viruses. 2025. PMID: 40284985 Free PMC article.
Abstract
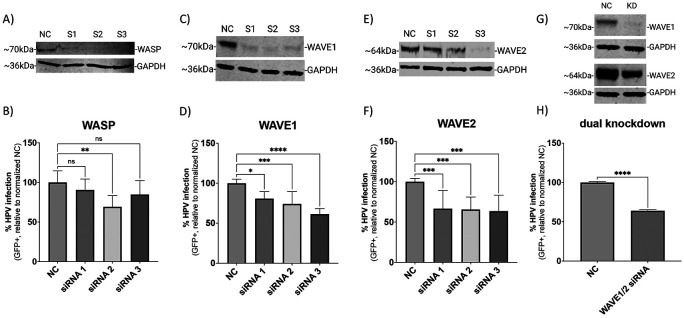
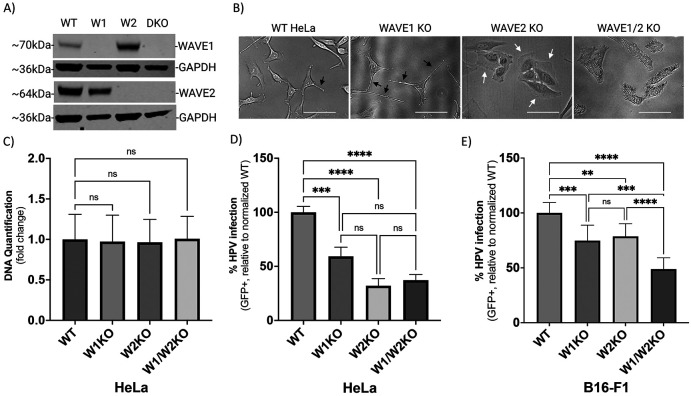
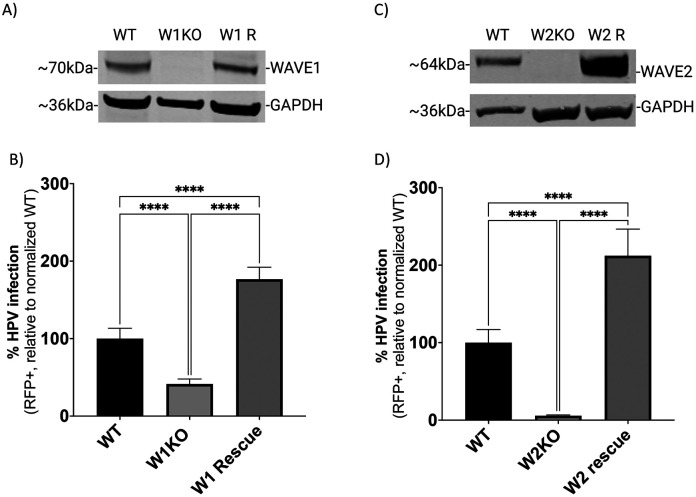
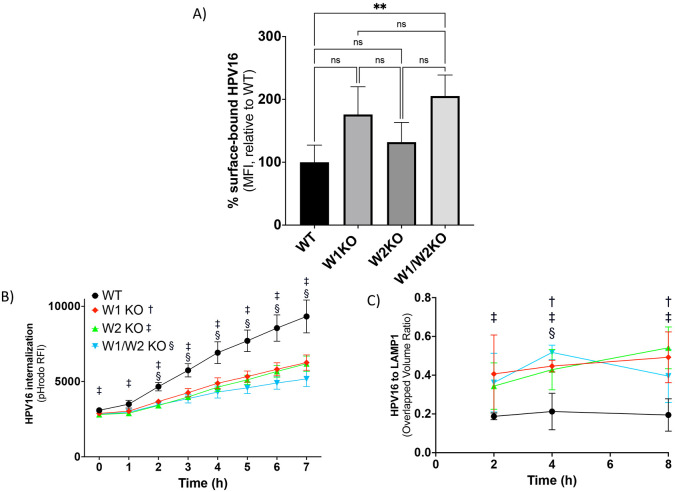
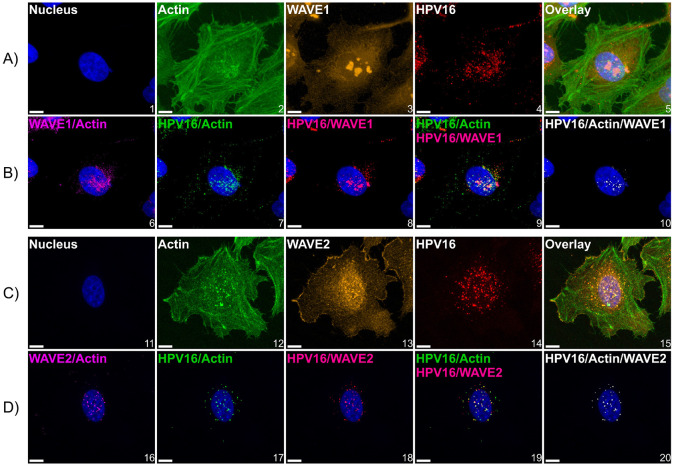
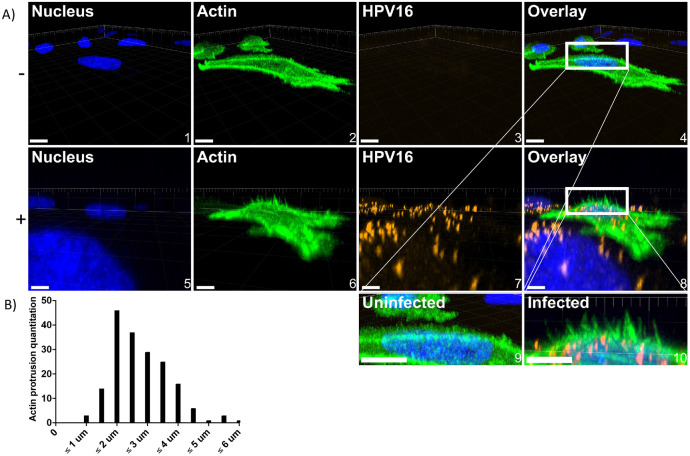
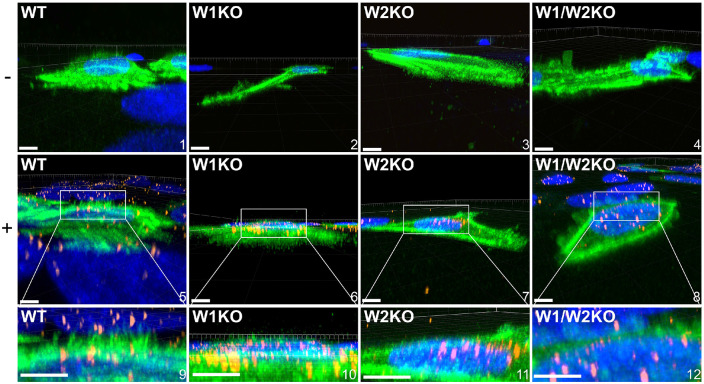
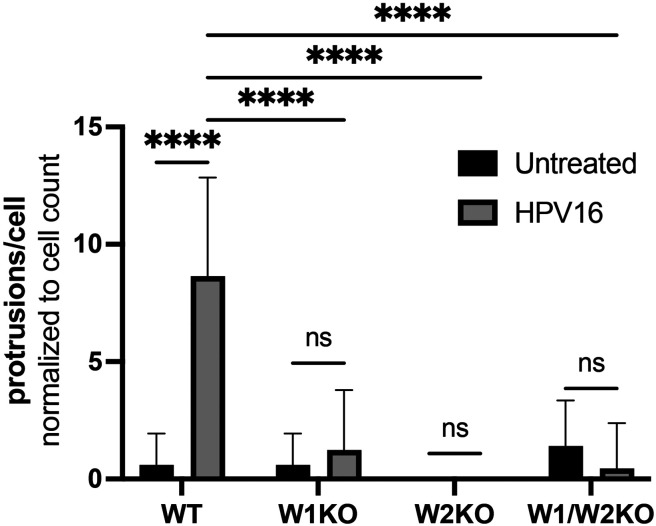
Human PapillomavirusType 16 (HPV16) is an etiological agent of human cancers that requires endocytosis to initiate infection. HPV16 entry into epithelial cells occurs through a non-canonical endocytic pathway that is actin-driven, but it is not well understood how HPV16-cell surface interactions trigger actin reorganization in a way that facilitates entry. This study provides evidence that Wiskott-Aldrich syndrome protein family verprolin-homologous proteins 1 and 2 (WAVE1 and WAVE2) are molecular mediators of the actin polymerization that facilitates HPV endocytosis and intracellular trafficking. We demonstrate through post-transcriptional gene silencing and genome editing that WAVE1 and WAVE2 are critical for efficient HPV16 infection, and that restoration of each in knockout cells rescues HPV16 infection. Cells lacking WAVE1, WAVE2, or both, internalize HPV16 at a significantly reduced rate. Analysis of fluorescently labeled cells exposed to HPV16 and acquired by confocal fluorescence microscopy revealed that HPV16, WAVE1, WAVE2, and actin are all colocalized at the cellular dorsal surface. We also found that HPV16 stimulates WAVE1 and WAVE2-mediated cellular dorsal surface filopodia formation during the viral endocytic process. Taken together, this study provides evidence that the HPV endocytic process needed for infection is controlled by actin reorganization into filopodial protrusions and that this process is mediated by WAVE1 and WAVE2.
Figures
References
-
- de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health. 2020. Feb 1;8(2):e180–90. - PubMed
-
- Molano M, van den Brule A, Plummer M, Weiderpass E, Posso H, Arslan A, et al. Determinants of Clearance of Human Papillomavirus Infections in Colombian Women with Normal Cytology: A Population-based, 5-Year Follow-up Study. Am J Epidemiol. 2003. Sep 1;158(5):486–94. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources