

This is a preprint.
Improved pangenomic classification accuracy with chain statistics
- PMID: 39554056
- PMCID: PMC11565826
- DOI: 10.1101/2024.10.29.620953
Improved pangenomic classification accuracy with chain statistics
Abstract
Compressed full-text indexes enable efficient sequence classification against a pangenome or tree-of-life index. Past work on compressed-index classification used matching statistics or pseudo-matching lengths to capture the fine-grained co-linearity of exact matches. But these fail to capture coarse-grained information about whether seeds appear co-linearly in the reference. We present a novel approach that additionally obtains coarse-grained co-linearity ("chain") statistics. We do this without using a chaining algorithm, which would require superlinear time in the number of matches. We start with a collection of strings, avoiding the multiple-alignment step required by graph approaches. We rapidly compute multi-maximal unique matches (multi-MUMs) and identify BWT sub-runs that correspond to these multi-MUMs. From these, we select those that can be "tunneled," and mark these with the corresponding multi-MUM identifiers. This yields an -space index for a collection of sequences having a length- BWT consisting of maximal equal-character runs. Using the index, we simultaneously compute fine-grained matching statistics and coarse-grained chain statistics in linear time with respect to query length. We found that this substantially improves classification accuracy compared to past compressed-indexing approaches and reaches the same level of accuracy as less efficient alignment-based methods.
Keywords: Pangenomics; sequence classification; text indexing.
Conflict of interest statement
Disclosure of Interests. Ben Langmead is the founder of InOrder Labs, LLC.
Figures

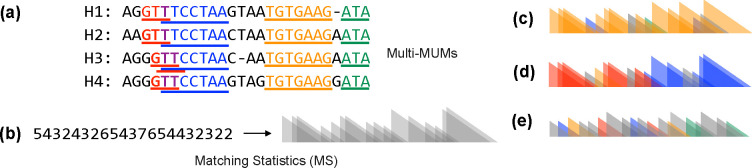
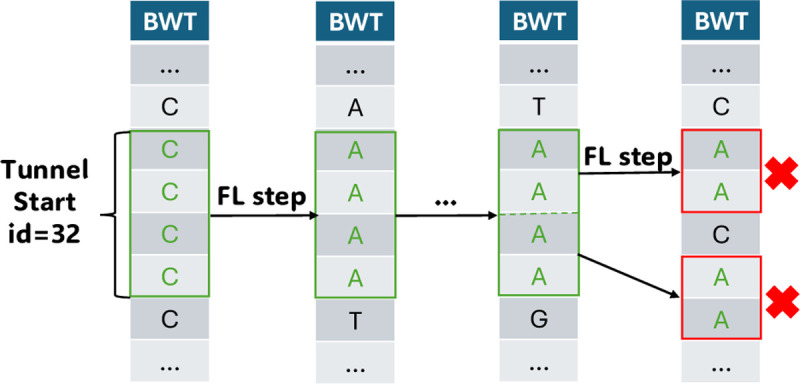


References
-
- Baier U.: On undetected redundancy in the burrows-wheeler transform. In: Annual Symposium on Combinatorial Pattern Matching (CPM 2018). Schloss Dagstuhl-Leibniz-Zentrum fuer Informatik; (2018)
-
- Baier U., Dede K.: Bwt tunnel planning is hard but manageable. 2019 Data Compression Conference (DCC) pp. 142–151 (2019), https://api.semanticscholar.org/CorpusID:155107876
-
- Baláž A., Gagie T., Goga A., Heumos S., Navarro G., Petescia A., Sirén J.: Wheeler maps. In: Latin American Symposium on Theoretical Informatics. pp. 178–192. Springer; (2024)
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous