

This is a preprint.
Integer programming framework for pangenome-based genome inference
- PMID: 39554168
- PMCID: PMC11565907
- DOI: 10.1101/2024.10.27.620212
Integer programming framework for pangenome-based genome inference
Update in
-
Pangenome-based genome inference using integer programming.Genome Res. 2025 Dec 3;35(12):2661-2670. doi: 10.1101/gr.280567.125. Genome Res. 2025. PMID: 40841174
Abstract
Affordable genotyping methods are essential in genomics. Commonly used genotyping methods primarily support single nucleotide variants and short indels but neglect structural variants. Additionally, accuracy of read alignments to a reference genome is unreliable in highly polymorphic and repetitive regions, further impacting genotyping performance. Recent works highlight the advantage of haplotype-resolved pangenome graphs in addressing these challenges. Building on these developments, we propose a rigorous alignment-free genotyping framework. Our formulation seeks a path through the pangenome graph that maximizes the matches between the path and substrings of sequencing reads (e.g., k-mers) while minimizing recombination events (haplotype switches) along the path. We prove that this problem is NP-Hard and develop efficient integer-programming solutions. We benchmarked the algorithm using downsampled short-read datasets from homozygous human cell lines with coverage ranging from 0.1× to 10×. Our algorithm accurately estimates complete major histocompatibility complex (MHC) haplotype sequences with small edit distances from the ground-truth sequences, providing a significant advantage over existing methods on low-coverage inputs. Although our algorithm is designed for haploid samples, we discuss future extensions to diploid samples.
Figures

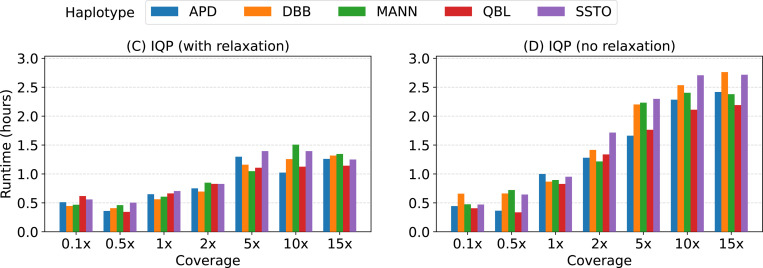


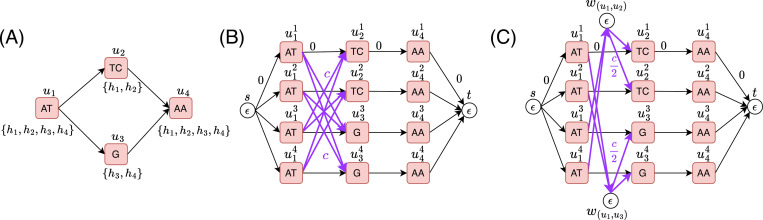
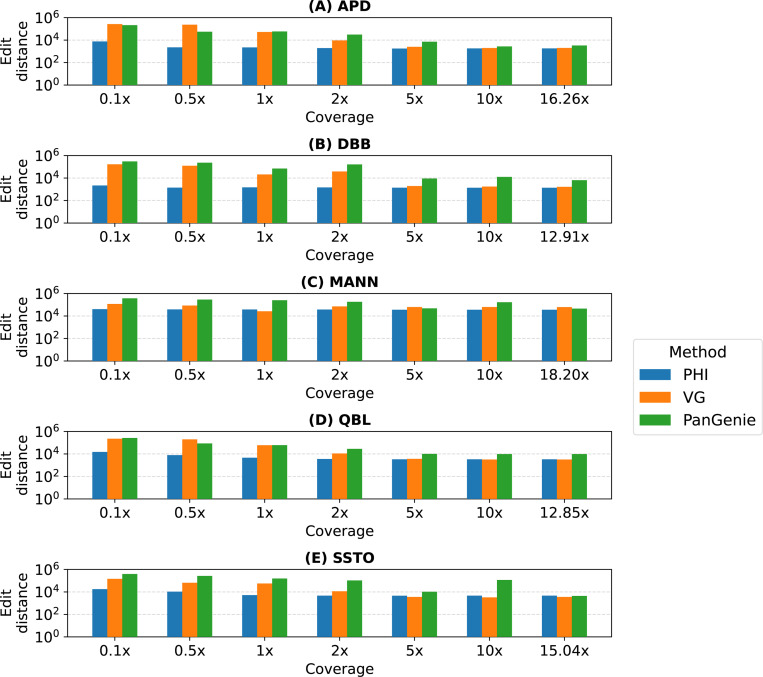

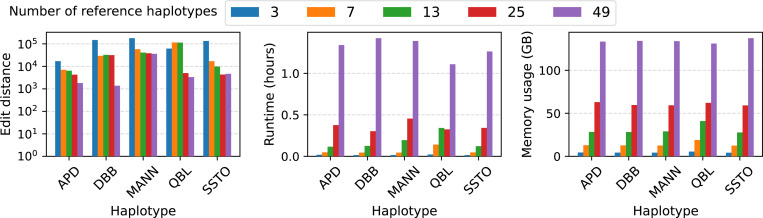
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous