This is a preprint.
GENCODE: massively expanding the lncRNA catalog through capture long-read RNA sequencing
- PMID: 39554180
- PMCID: PMC11565817
- DOI: 10.1101/2024.10.29.620654
GENCODE: massively expanding the lncRNA catalog through capture long-read RNA sequencing
Abstract
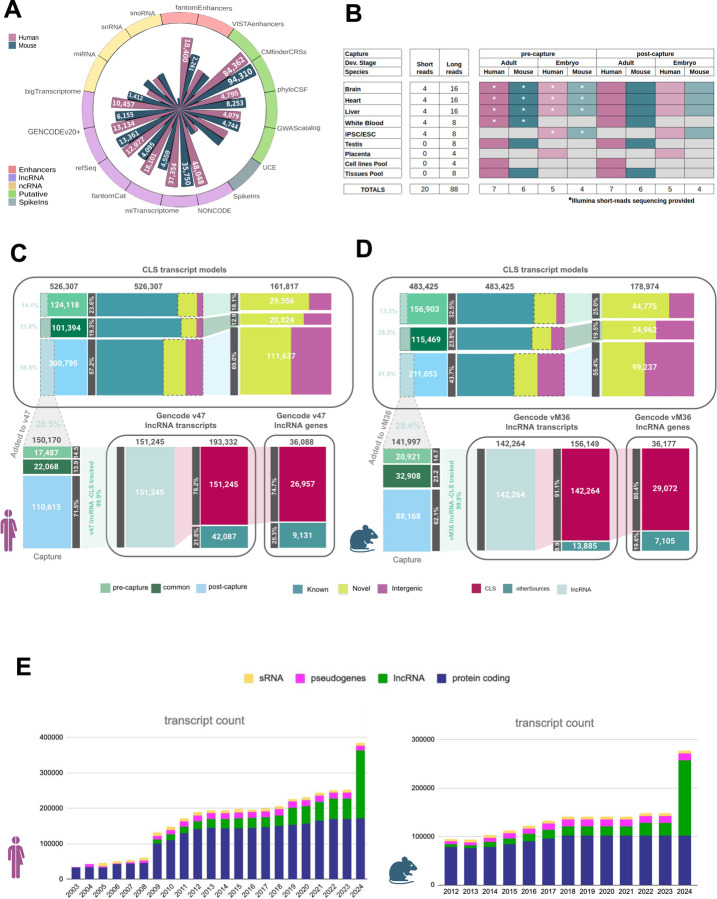
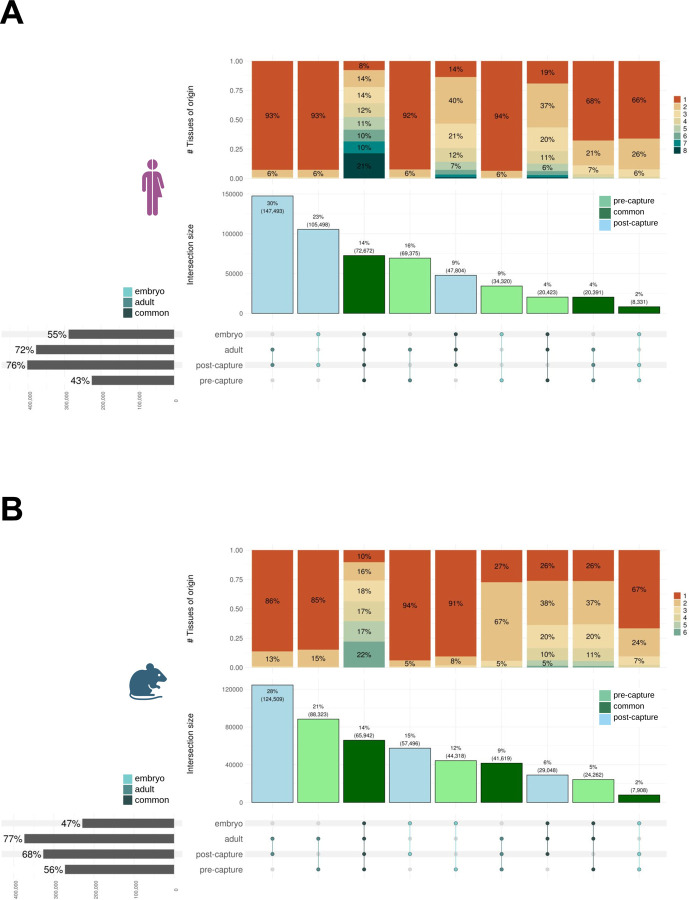
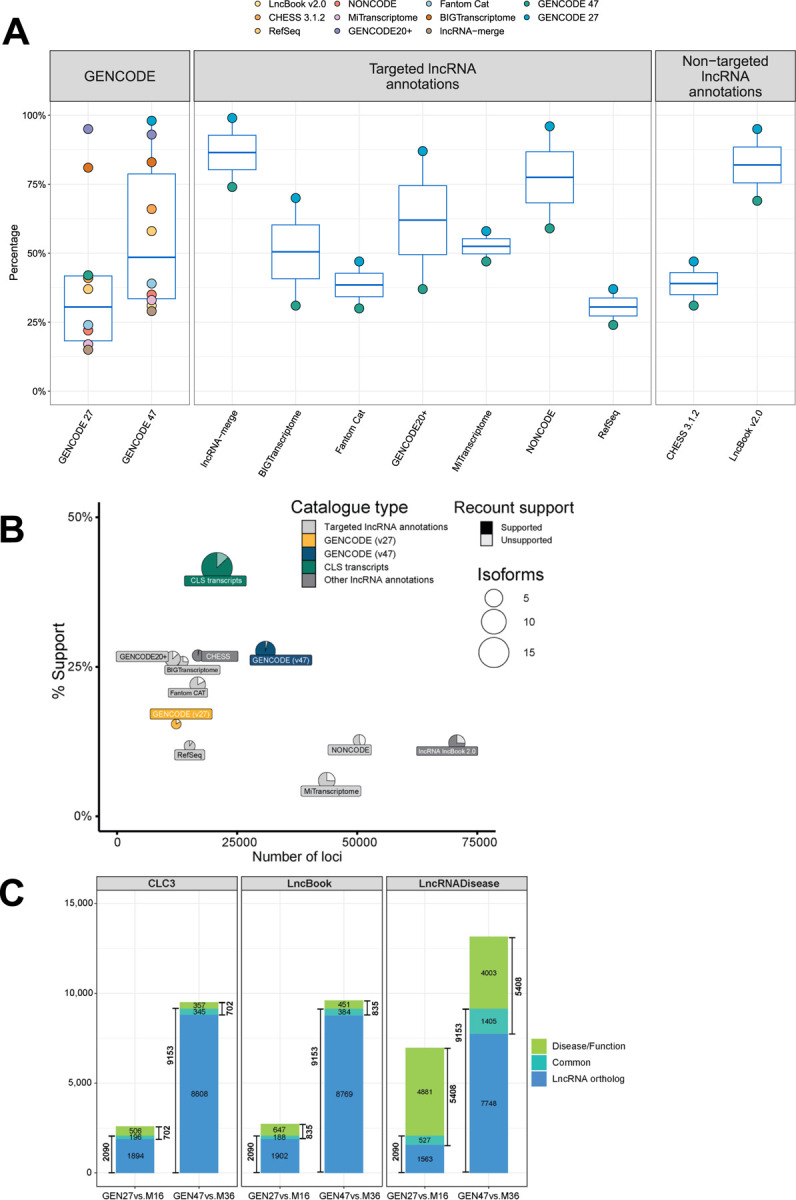
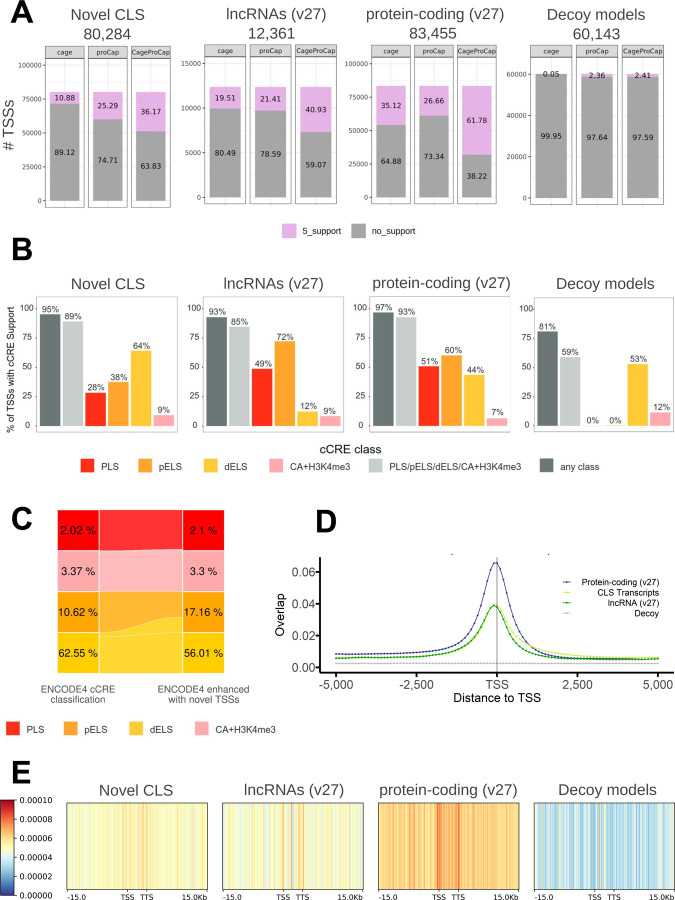
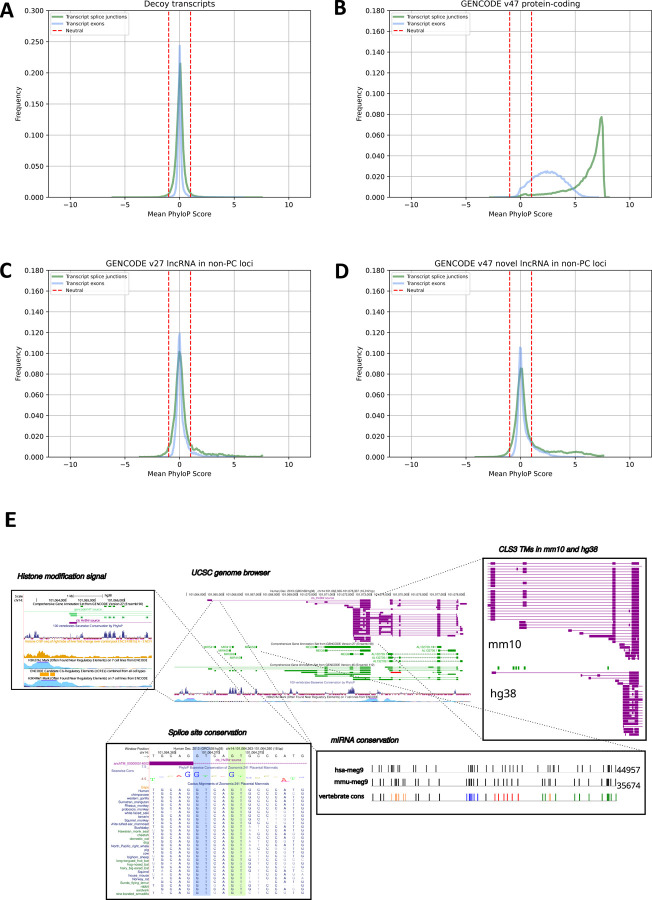
Accurate and complete gene annotations are indispensable for understanding how genome sequences encode biological functions. For twenty years, the GENCODE consortium has developed reference annotations for the human and mouse genomes, becoming a foundation for biomedical and genomics communities worldwide. Nevertheless, collections of important yet poorly-understood gene classes like long non-coding RNAs (lncRNAs) remain incomplete and scattered across multiple, uncoordinated catalogs, slowing down progress in the field. To address these issues, GENCODE has undertaken the most comprehensive lncRNAs annotation effort to date. This is founded on the manual annotation of full-length targeted long-read sequencing, on matched embryonic and adult tissues, of orthologous regions in human and mouse. Altogether 17,931 novel human genes (140,268 novel transcripts) and 22,784 novel mouse genes (136,169 novel transcripts) have been added to the GENCODE catalog representing a 2-fold and 6-fold increase in transcripts, respectively - the greatest increase since the sequencing of the human genome. Novel gene annotations display evolutionary constraints, have well-formed promoter regions, and link to phenotype-associated genetic variants. They greatly enhance the functional interpretability of the human genome, as they help explain millions of previously-mapped "orphan" omics measurements corresponding to transcription start sites, chromatin modifications and transcription factor binding sites. Crucially, our targeted design assigned human-mouse orthologs at a rate beyond previous studies, tripling the number of human disease-associated lncRNAs with mouse orthologs. The expanded and enhanced GENCODE lncRNA annotations mark a critical step towards deciphering the human and mouse genomes.
Conflict of interest statement
Competing Interests The authors declare no competing interests.
Figures