

This is a preprint.
CCAFE: Estimating Case and Control Allele Frequencies from GWAS Summary Statistics
- PMID: 39554201
- PMCID: PMC11565872
- DOI: 10.1101/2024.10.24.619530
CCAFE: Estimating Case and Control Allele Frequencies from GWAS Summary Statistics
Abstract
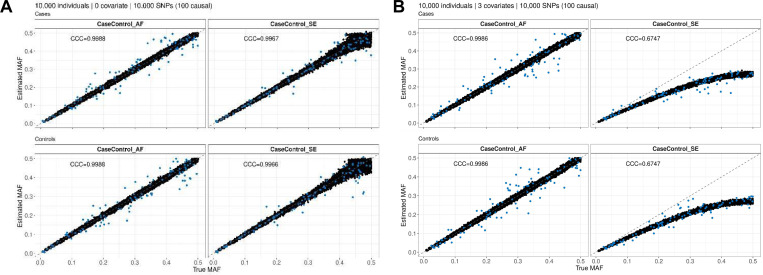
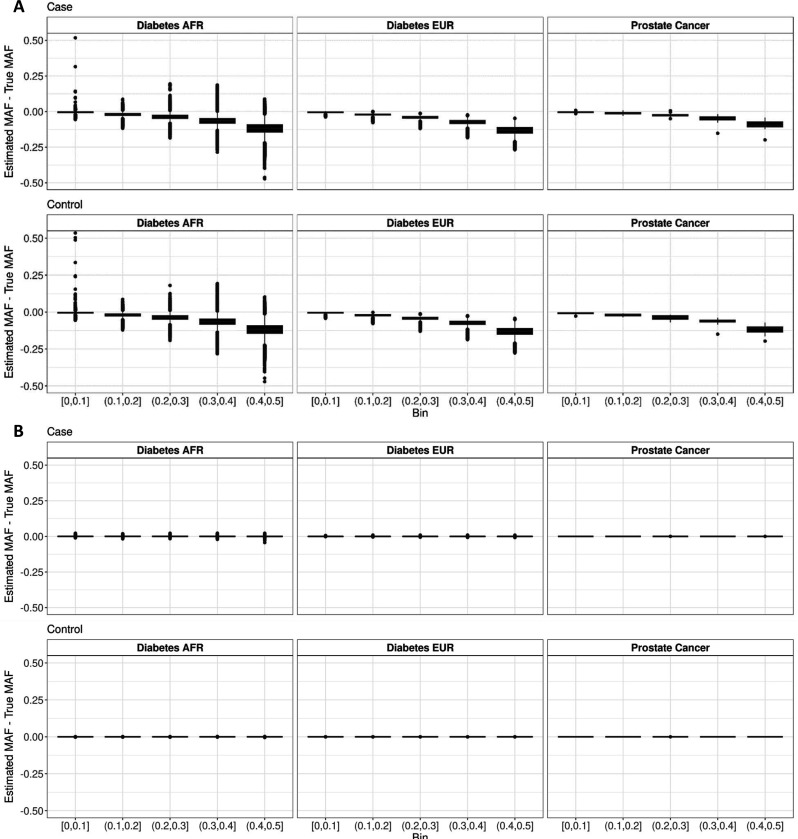
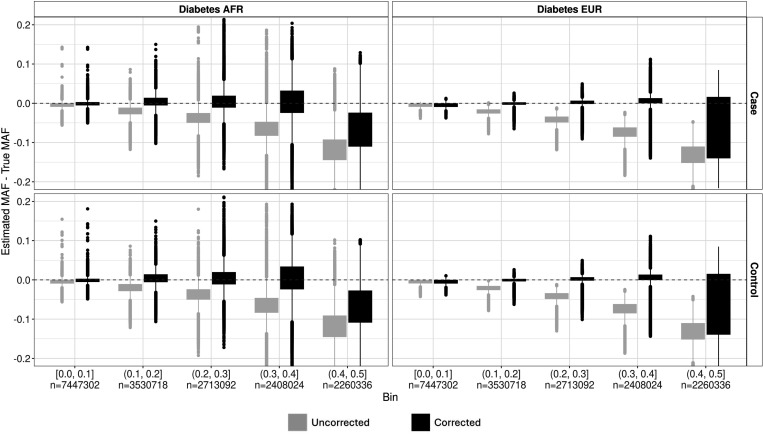
Methods involving summary statistics in genetics can be quite powerful but can be limited in utility. For instance, many post-hoc analyses of disease studies require case and control allele frequencies (AFs), which are not always published. We present two frameworks to derive case and control AFs from GWAS summary statistics using the odds ratio, case and control sample sizes, and either the total (case and control aggregated) AF or standard error (SE). In simulations and real data, derivations of case and controls AFs using total AF is highly accurate across all settings (e.g., minor AF, condition prevalence). Conversely, derivations using SE underestimate common variant AFs (e.g. minor allele frequency >0.3) in the presence of covariates. We develop an adjustment using gnomAD AFs as a proxy for true AFs, which reduces the bias when using SE. While estimating case and control AFs using the total AF is preferred due to its high accuracy, estimating from the SE can be used more broadly since SE can be derived from p-values and beta estimates, which are commonly provided. The methods provided here expand the utility of publicly available genetic summary statistics and promote the reusability of genomic data. The R package CCAFE, with implementations of both methods, is freely available on Bioconductor and GitHub.
Keywords: GWAS; R package; allele frequencies; statistical genetics; summary statistics.
Figures
References
-
- Buniello A., MacArthur J. A. L., Cerezo M., Harris L. W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E., Suveges D., Vrousgou O., Whetzel P. L., Amode R., Guillen J. A., Riat H. S., Trevanion S. J., Hall P., Junkins H., Flicek P., Burdett T., Hindorff L. A., Cunningham F., & Parkinson H. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res, 47(D1), D1005–D1012. 10.1093/nar/gky1120 - DOI - PMC - PubMed
-
- Hayhurst J., Buniello A., Harris L., Mosaku A., Chang C., Gignoux C. R., Hatzikotoulas K., Karim M. A., Lambert S. A., Lyon M., McMahon A., Okada Y., Pirastu N., Rayner N. W., Schwartzentruber J., Vaughan R., Verma S., Wilder S. P., Cunningham F., Hindorff L., Wiley K., Parkinson H., & Barroso I. (2023). A community driven GWAS summary statistics standard. bioRxiv.
-
- Hendricks A. E., Billups S. C., Pike H. N. C., Farooqi I. S., Zeggini E., Santorico S. A., Barroso I., & Dupuis J. (2018). ProxECAT: Proxy External Controls Association Test. A new case-control gene region association test using allele frequencies from public controls. PLoS Genet, 14(10), e1007591. 10.1371/journal.pgen.1007591 - DOI - PMC - PubMed
-
- Hinrichs A. S., Karolchik D., Baertsch R., Barber G. P., Bejerano G., Clawson H., Diekhans M., Furey T. S., Harte R. A., Hsu F., Hillman-Jackson J., Kuhn R. M., Pedersen J. S., Pohl A., Raney B. J., Rosenbloom K. R., Siepel A., Smith K. E., Sugnet C. W., Sultan-Qurraie A., Thomas D. J., Trumbower H., Weber R. J., Weirauch M., Zweig A. S., Haussler D., & Kent W. J. (2006). The UCSC Genome Browser Database: update 2006. Nucleic Acids Res, 34(Database issue), D590–598. 10.1093/nar/gkj144 - DOI - PMC - PubMed
-
- Huber W., Carey V. J., Gentleman R., Anders S., Carlson M., Carvalho B. S., Bravo H. C., Davis S., Gatto L., Girke T., Gottardo R., Hahne F., Hansen K. D., Irizarry R. A., Lawrence M., Love M. I., MacDonald J., Obenchain V., Oles A. K., Pages H., Reyes A., Shannon P., Smyth G. K., Tenenbaum D., Waldron L., & Morgan M. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods, 12(2), 115–121. 10.1038/nmeth.3252 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources