

Probe set selection for targeted spatial transcriptomics
- PMID: 39558096
- PMCID: PMC11621025
- DOI: 10.1038/s41592-024-02496-z
Probe set selection for targeted spatial transcriptomics
Abstract
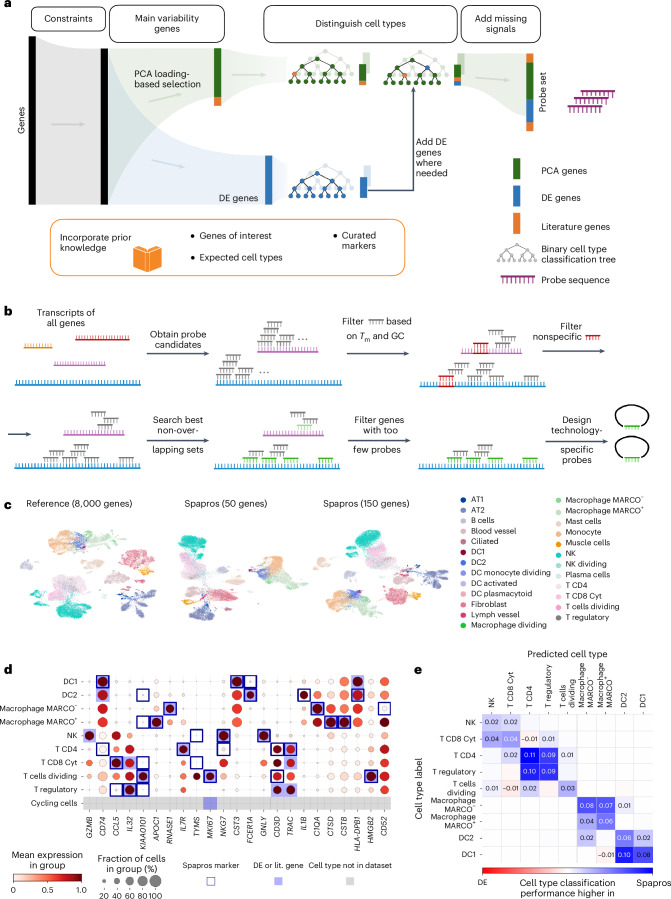
Targeted spatial transcriptomic methods capture the topology of cell types and states in tissues at single-cell and subcellular resolution by measuring the expression of a predefined set of genes. The selection of an optimal set of probed genes is crucial for capturing the spatial signals present in a tissue. This requires selecting the most informative, yet minimal, set of genes to profile (gene set selection) for which it is possible to build probes (probe design). However, current selections often rely on marker genes, precluding them from detecting continuous spatial signals or new states. We present Spapros, an end-to-end probe set selection pipeline that optimizes both gene set specificity for cell type identification and within-cell type expression variation to resolve spatially distinct populations while considering prior knowledge as well as probe design and expression constraints. We evaluated Spapros and show that it outperforms other selection approaches in both cell type recovery and recovering expression variation beyond cell types. Furthermore, we used Spapros to design a single-cell resolution in situ hybridization on tissues (SCRINSHOT) experiment of adult lung tissue to demonstrate how probes selected with Spapros identify cell types of interest and detect spatial variation even within cell types.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: F.J.T. consults for Immunai Inc., CytoReason Ltd, Cellarity and BioTuring Inc., and has an ownership interest in Dermagnostix GmbH and Cellarity. M.D.L. contracted for the Chan Zuckerberg Initiative and received speaker fees from Pfizer and Janssen Pharmaceuticals. P.H., F.K. and T.B. acknowledge support from TKP2021-EGA09, Horizon-BIALYMPH, -SYMMETRY, -SWEEPICS, -Fair-CHARM and OTKA-SNN 139455.
Figures













References
-
- Asp, M. et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell179, 1647–1660 (2019). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
