

Towards routine proteome profiling of FFPE tissue: insights from a 1,220-case pan-cancer study
- PMID: 39558110
- PMCID: PMC11697351
- DOI: 10.1038/s44318-024-00289-w
Towards routine proteome profiling of FFPE tissue: insights from a 1,220-case pan-cancer study
Abstract
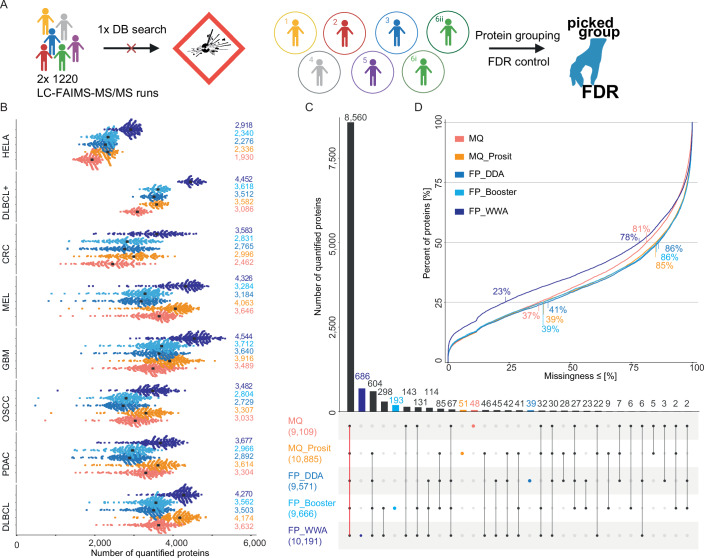
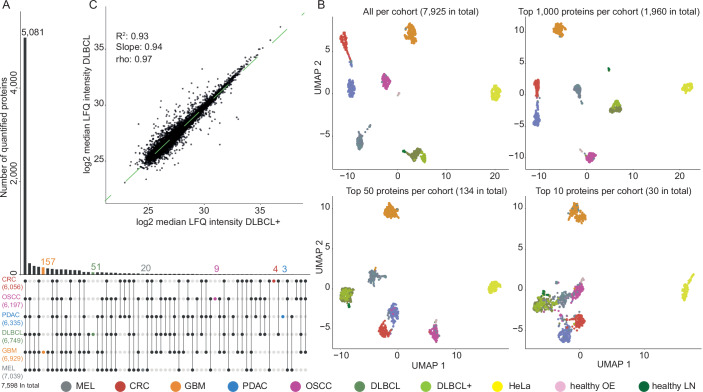
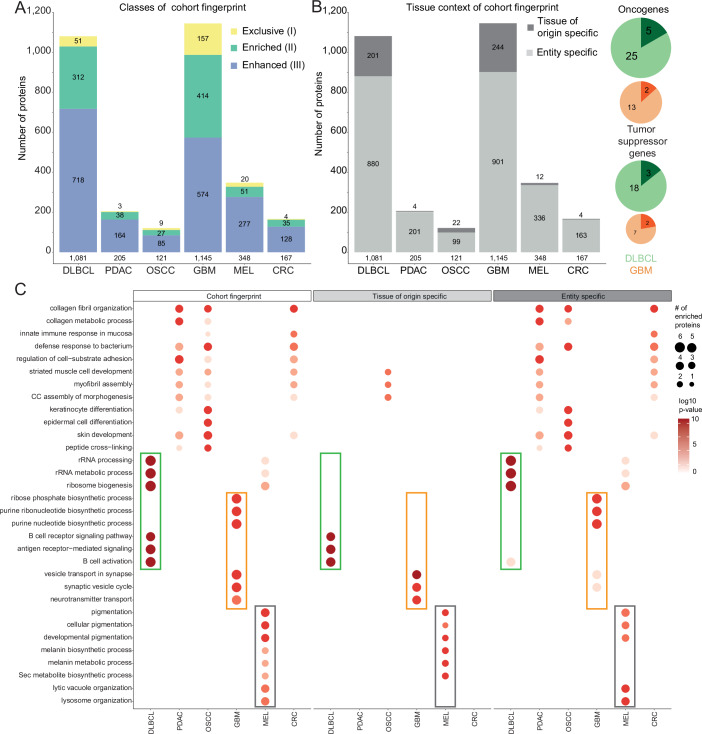
Proteome profiling of formalin-fixed paraffin-embedded (FFPE) specimens has gained traction for the analysis of cancer tissue for the discovery of molecular biomarkers. However, reports so far focused on single cancer entities, comprised relatively few cases and did not assess the long-term performance of experimental workflows. In this study, we analyze 1220 tumors from six cancer entities processed over the course of three years. Key findings include the need for a new normalization method ensuring equal and reproducible sample loading for LC-MS/MS analysis across cohorts, showing that tumors can, on average, be profiled to a depth of >4000 proteins and discovering that current software fails to process such large ion mobility-based online fractionated datasets. We report the first comprehensive pan-cancer proteome expression resource for FFPE material comprising 11,000 proteins which is of immediate utility to the scientific community, and can be explored via a web resource. It enables a range of analyses including quantitative comparisons of proteins between patients and cohorts, the discovery of protein fingerprints representing the tissue of origin or proteins enriched in certain cancer entities.
Keywords: Clinical Proteomics; Mass Spectrometry; Pan-cancer; Public Pan-cancer FFPE Resource; TIC Normalization.
© 2024. The Author(s).
Conflict of interest statement
Disclosure and competing interests statement. BK is the founder and shareholder of OmicScouts and MSAID. He has no operational role in either company. The remaining authors declare no competing interests.
Figures








References
-
- Bhatia HS, Brunner AD, Öztürk F, Kapoor S, Rong Z, Mai H, Thielert M, Ali M, Al-Maskari R, Paetzold JC et al (2022) Spatial proteomics in three-dimensional intact specimens. Cell 185:5040–5058.e5019 - PubMed
-
- Buczak K, Kirkpatrick JM, Truckenmueller F, Santinha D, Ferreira L, Roessler S, Singer S, Beck M, Ori A (2020) Spatially resolved analysis of FFPE tissue proteomes by quantitative mass spectrometry. Nat Protoc 15:2956–2979 - PubMed
-
- Coscia F, Doll S, Bech JM, Schweizer L, Mund A, Lengyel E, Lindebjerg J, Madsen GI, Moreira JM, Mann M (2020) A streamlined mass spectrometry-based proteomics workflow for large-scale FFPE tissue analysis. J Pathol 251:100–112 - PubMed
-
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
