

Small-molecule disruption of androgen receptor-dependent chromatin clusters
- PMID: 39560645
- PMCID: PMC11621760
- DOI: 10.1073/pnas.2406239121
Small-molecule disruption of androgen receptor-dependent chromatin clusters
Abstract
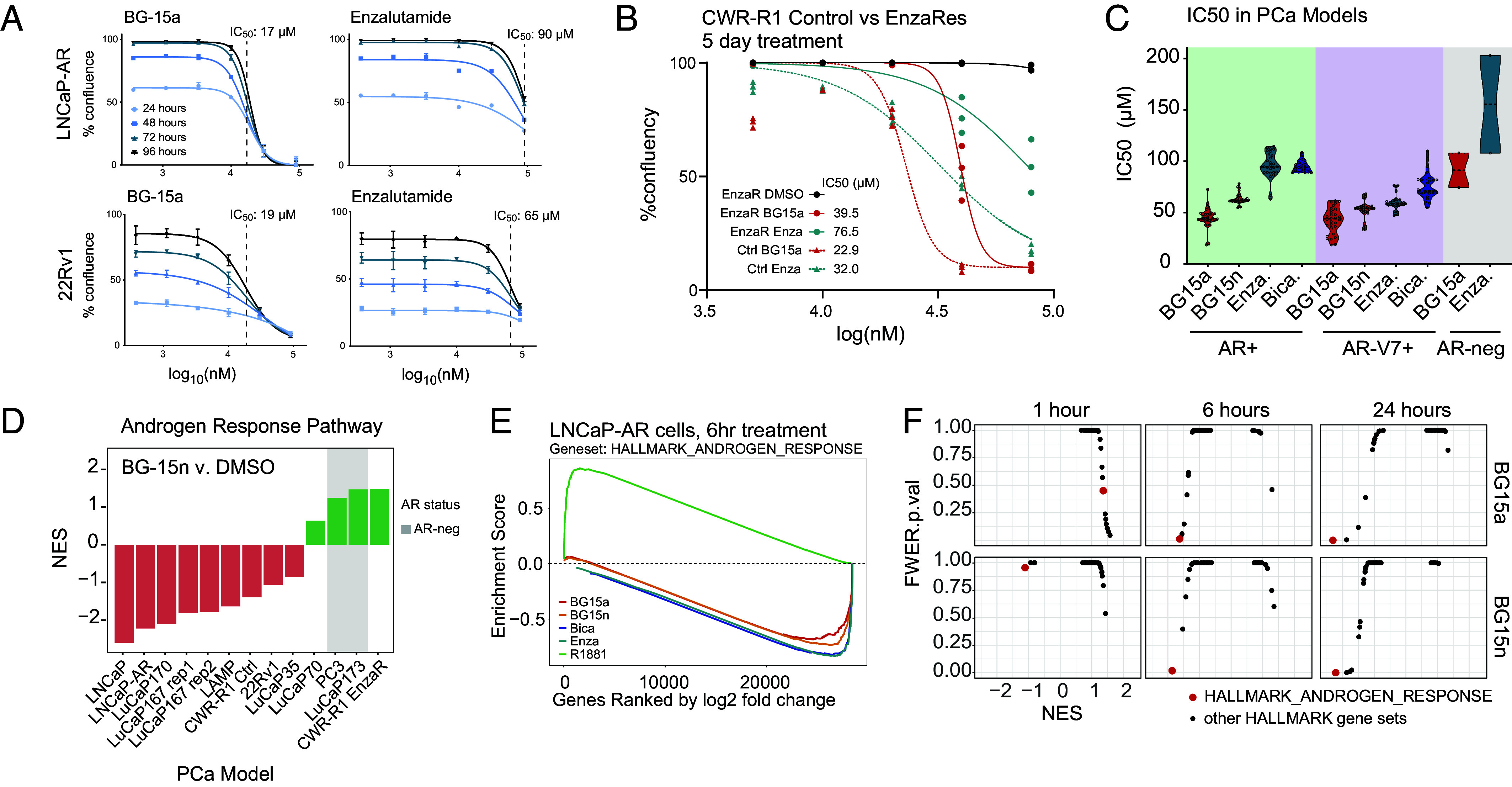
Sustained androgen receptor (AR) signaling during relapse is a central driver of metastatic castration-resistant prostate cancer (mCRPC). Current AR antagonists, such as enzalutamide, fail to provide long-term benefit for the mCRPC patients who have dramatic increases in AR expression. Here, we report AR antagonists with efficacy in AR-overexpressing models. These molecules bind to the ligand-binding domain of the AR, promote AR localization to the nucleus, yet potently and selectively down-regulate AR-target genes. The molecules BG-15a and the pharmacokinetically optimized BG-15n elicit a decrease in cell and tumor growth in vitro and in vivo in models of mCRPC. BG-15a/n treatment causes the collapse of chromatin loops between enhancers and promoters at key genes in the AR-driven epigenome. AR binding in the promoter, as well as 3D chromatin clustering, is needed for genes to respond. BG-15a/n represent promising agents for treating patients with relapsed AR-driven mCRPC tumors.
Keywords: androgen receptor; chromatin architecture; epigenetics; gene regulation; prostate cancer.
Conflict of interest statement
Competing interests statement:E.C. received research funding under institutional SRA from Janssen Research and Development, Bayer Pharmaceuticals, KronosBio, Forma Pharmaceutics Foghorn, Gilead, Sanofi, AbbVie, MacrogGenics, Astra Zeneca, GSK, and K36. B.E.G., A.K.O., S.T., W.D.F., and J.S. are co-inventors on a patent covering this technology (WO2021150603A1). P.S.N. receives personal fees from Janssen, Bristol Myers Squibb, Pfizer, and Merck and grants from Janssen. The other authors declare no competing interests.
Figures





References
-
- Pirtskhalaishvili G., Hrebinko R. L., Nelson J. B., The treatment of prostate cancer—An overview of current options. Cancer Pract. 9, 295–306 (2001). - PubMed
MeSH terms
Substances
Grants and funding
- W81XWH-19-1-0715/U.S. Department of Defense (DOD)
- P50 CA097186/CA/NCI NIH HHS/United States
- CA097186/Prostate Cancer Foundation (PCF)
- 5R01CA291963-02/HHS | National Institutes of Health (NIH)
- K22 CA255594/CA/NCI NIH HHS/United States
- P50CA97186/HHS | National Institutes of Health (NIH)
- 1K22CA255594/HHS | National Institutes of Health (NIH)
- RO1 CA266013/HHS | National Institutes of Health (NIH)
- R01 CA266013/CA/NCI NIH HHS/United States
- S10 OD024996/OD/NIH HHS/United States
- R01 CA291963/CA/NCI NIH HHS/United States
- P01 CA163227/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
