

Preclinical evaluation of antigen-sensitive B7-H3-targeting nanobody-based CAR-T cells in glioblastoma cautions for on-target, off-tumor toxicity
- PMID: 39562005
- PMCID: PMC11575280
- DOI: 10.1136/jitc-2024-009110
Preclinical evaluation of antigen-sensitive B7-H3-targeting nanobody-based CAR-T cells in glioblastoma cautions for on-target, off-tumor toxicity
Abstract
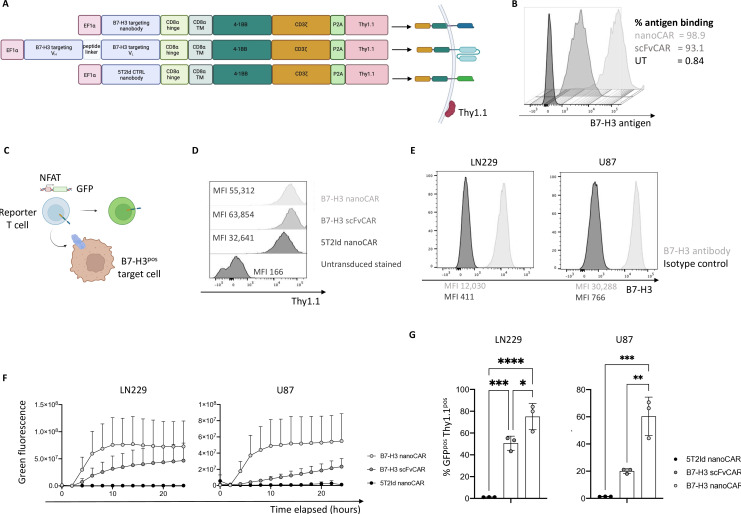
Background: Glioblastoma is the most common lethal primary brain tumor, urging evaluation of new treatment options. Chimeric antigen receptor (CAR)-T cells targeting B7 homolog 3 (B7-H3) are promising because of the overexpression of B7-H3 on glioblastoma cells but not on healthy brain tissue. Nanobody-based (nano)CARs are gaining increasing attention as promising alternatives to classical single-chain variable fragment-based (scFv)CARs, because of their single-domain nature and low immunogenicity. Still, B7-H3 nanoCAR-T cells have not been extensively studied in glioblastoma.
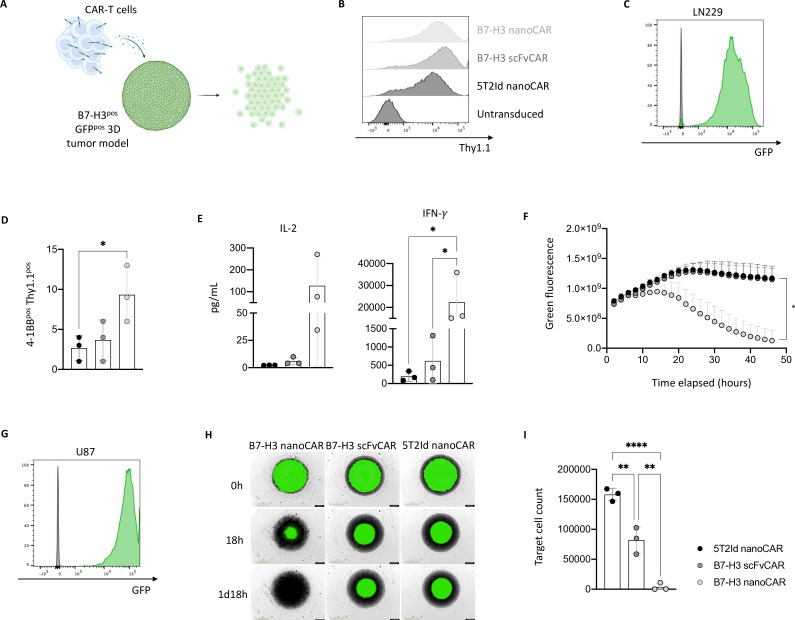
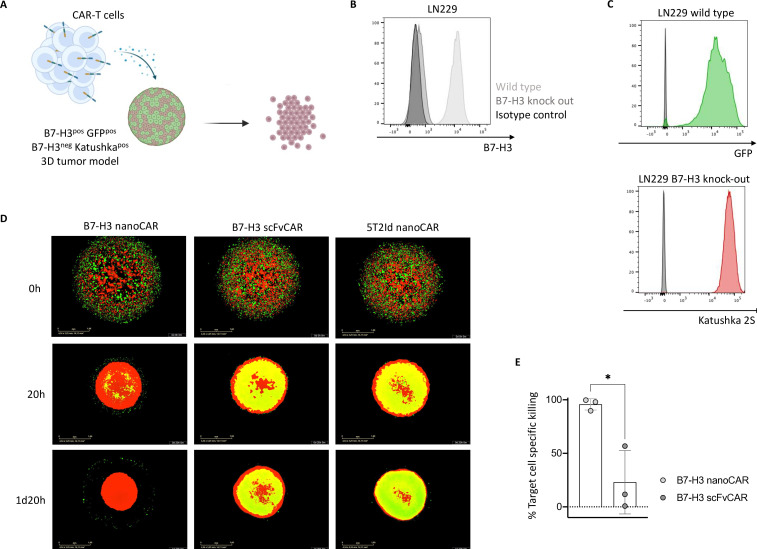
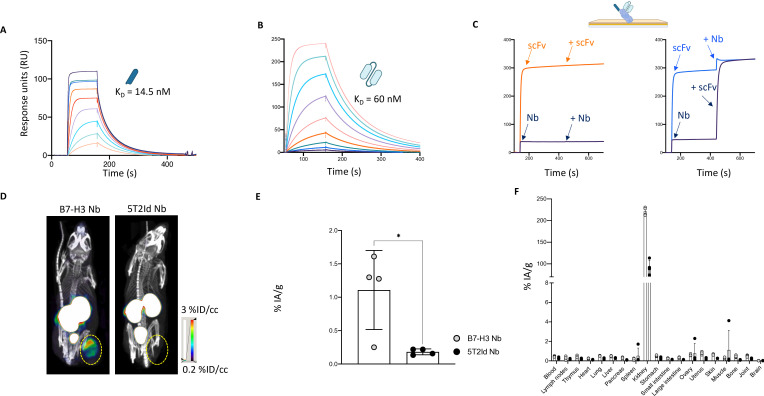
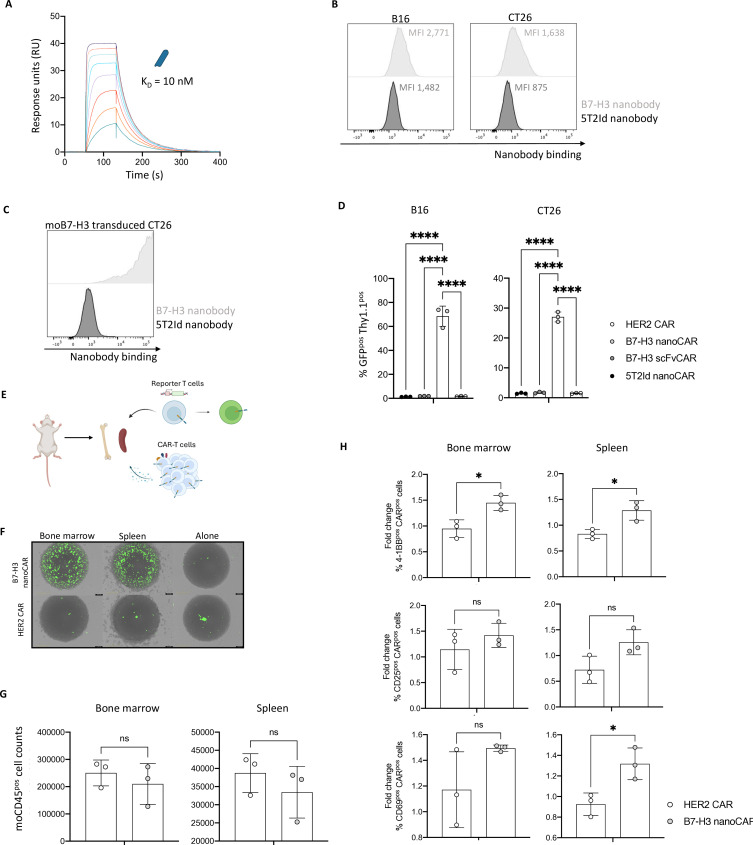
Methods: B7-H3 nanoCAR- and scFvCAR-T cells were developed and evaluated in human glioblastoma models. NanoCAR-T cells targeting an irrelevant antigen served as control. T cell activation, cytokine secretion and killing capacity were evaluated in vitro using ELISA, live cell imaging and flow cytometry. Antigen-specific killing was assessed by generating B7-H3 knock-out cells using Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-genome editing. The tumor tracing capacity of the B7-H3 nanobody was first evaluated in vivo using nuclear imaging. Then, the therapeutic potential of the nanoCAR-T cells was evaluated in a xenograft glioblastoma model.
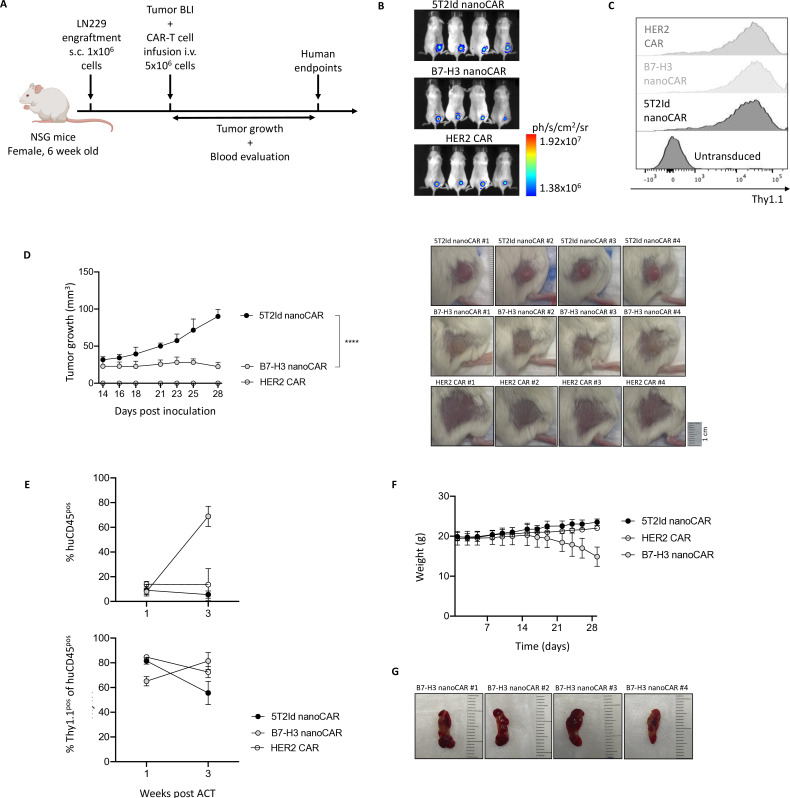
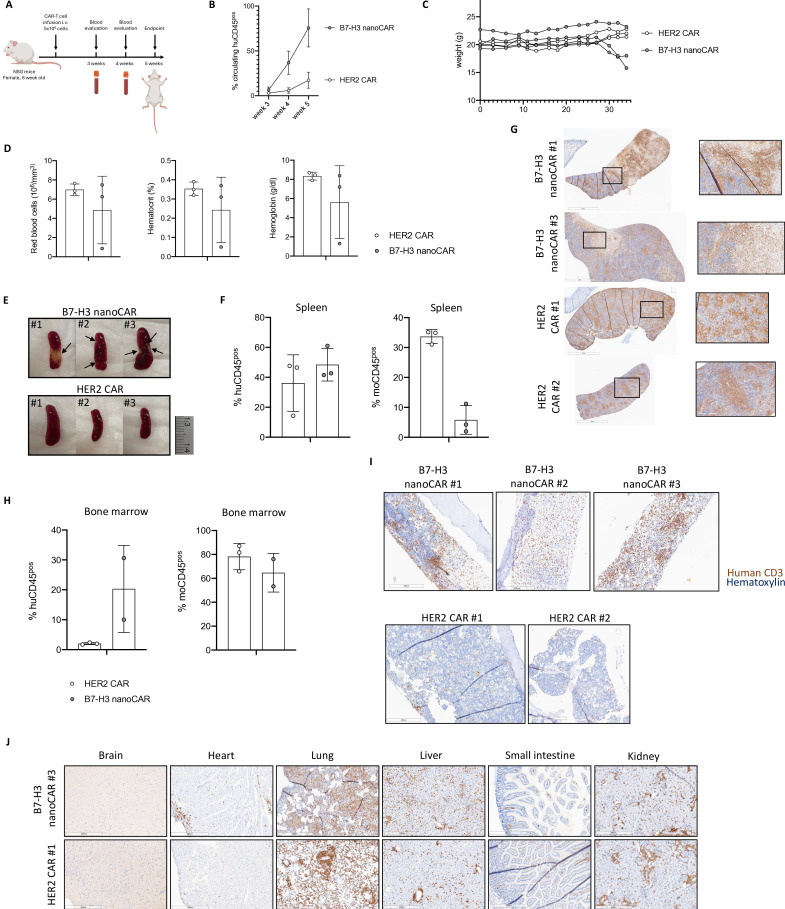
Results: We showed that B7-H3 nanoCAR-T cells were most efficient in lysing B7-H3pos glioblastoma cells in vitro. Lack of glioblastoma killing by control nanoCAR-T cells and lack of B7-H3neg glioblastoma killing by B7-H3 nanoCAR-T cells showed antigen-specificity. We showed in vivo tumor targeting capacity of the B7-H3 nanobody-used for the nanoCAR design-in nuclear imaging experiments. Evaluation of the nanoCAR-T cells in vivo showed tumor control in mice treated with B7-H3 nanoCAR-T cells in contrast to progressive disease in mice treated with control nanoCAR-T cells. However, we observed limiting toxicity in mice treated with B7-H3 nanoCAR-T cells and showed that the B7-H3 nanoCAR-T cells are activated even by low levels of mouse B7-H3 expression.
Conclusions: B7-H3 nanoCAR-T cells showed promise for glioblastoma therapy following in vitro characterization, but limiting in vivo toxicity was observed. Off-tumor recognition of healthy mouse tissue by the cross-reactive B7-H3 nanoCAR-T cells was identified as a potential cause for this toxicity, warranting caution when using highly sensitive nanoCAR-T cells, recognizing the low-level expression of B7-H3 on healthy tissue.
Keywords: Adoptive cell therapy - ACT; Chimeric antigen receptor - CAR; Immunotherapy; Solid tumor; T cell.
© Author(s) (or their employer(s)) 2024. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: No, there are no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials