

A systematic quantitative approach comprehensively defines domain-specific functional pathways linked to Schizosaccharomyces pombe heterochromatin regulation
- PMID: 39565189
- PMCID: PMC11662645
- DOI: 10.1093/nar/gkae1024
A systematic quantitative approach comprehensively defines domain-specific functional pathways linked to Schizosaccharomyces pombe heterochromatin regulation
Abstract
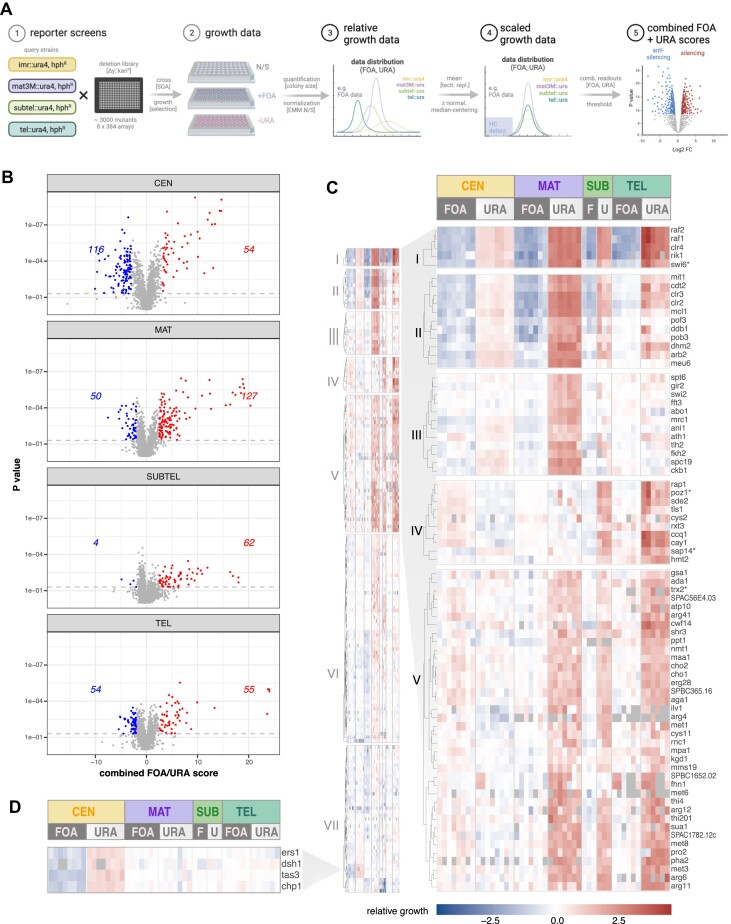
Heterochromatin plays a critical role in regulating gene expression and maintaining genome integrity. While structural and enzymatic components have been linked to heterochromatin establishment, a comprehensive view of the underlying pathways at diverse heterochromatin domains remains elusive. Here, we developed a systematic approach to identify factors involved in heterochromatin silencing at pericentromeres, subtelomeres and the silent mating type locus in Schizosaccharomyces pombe. Using quantitative measures, iterative genetic screening and domain-specific heterochromatin reporters, we identified 369 mutants with different degrees of reduced or enhanced silencing. As expected, mutations in the core heterochromatin machinery globally decreased silencing. However, most other mutants exhibited distinct qualitative and quantitative profiles that indicate heterochromatin domain-specific functions, as seen for example for metabolic pathways affecting primarily subtelomere silencing. Moreover, similar phenotypic profiles revealed shared functions for subunits within complexes. We further discovered that the uncharacterized protein Dhm2 plays a crucial role in heterochromatin maintenance, affecting the inheritance of H3K9 methylation and the clonal propagation of the repressed state. Additionally, Dhm2 loss resulted in delayed S-phase progression and replication stress. Collectively, our systematic approach unveiled a landscape of domain-specific heterochromatin regulators controlling distinct states and identified Dhm2 as a previously unknown factor linked to heterochromatin inheritance and replication fidelity.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures






Update of
-
A systematic quantitative approach comprehensively defines domain-specific functional pathways linked to Schizosaccharomyces pombe heterochromatin regulation.bioRxiv [Preprint]. 2024 Feb 15:2024.02.13.579970. doi: 10.1101/2024.02.13.579970. bioRxiv. 2024. Update in: Nucleic Acids Res. 2024 Dec 11;52(22):13665-13689. doi: 10.1093/nar/gkae1024. PMID: 38405799 Free PMC article. Updated. Preprint.
Similar articles
-
A systematic quantitative approach comprehensively defines domain-specific functional pathways linked to Schizosaccharomyces pombe heterochromatin regulation.bioRxiv [Preprint]. 2024 Feb 15:2024.02.13.579970. doi: 10.1101/2024.02.13.579970. bioRxiv. 2024. Update in: Nucleic Acids Res. 2024 Dec 11;52(22):13665-13689. doi: 10.1093/nar/gkae1024. PMID: 38405799 Free PMC article. Updated. Preprint.
-
TOR complex 2 in fission yeast is required for chromatin-mediated gene silencing and assembly of heterochromatic domains at subtelomeres.J Biol Chem. 2018 May 25;293(21):8138-8150. doi: 10.1074/jbc.RA118.002270. Epub 2018 Apr 9. J Biol Chem. 2018. PMID: 29632066 Free PMC article.
-
Role for cohesin in the formation of a heterochromatic domain at fission yeast subtelomeres.Mol Cell Biol. 2011 Mar;31(5):1088-97. doi: 10.1128/MCB.01290-10. Epub 2010 Dec 28. Mol Cell Biol. 2011. PMID: 21189291 Free PMC article.
-
RNAi-mediated chromatin silencing in fission yeast.Curr Top Microbiol Immunol. 2008;320:157-83. doi: 10.1007/978-3-540-75157-1_8. Curr Top Microbiol Immunol. 2008. PMID: 18268844 Review.
-
The molecular basis of heterochromatin assembly and epigenetic inheritance.Mol Cell. 2023 Jun 1;83(11):1767-1785. doi: 10.1016/j.molcel.2023.04.020. Epub 2023 May 18. Mol Cell. 2023. PMID: 37207657 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
