

Gene count estimation with pytximport enables reproducible analysis of bulk RNA sequencing data in Python
- PMID: 39565903
- PMCID: PMC11629965
- DOI: 10.1093/bioinformatics/btae700
Gene count estimation with pytximport enables reproducible analysis of bulk RNA sequencing data in Python
Abstract
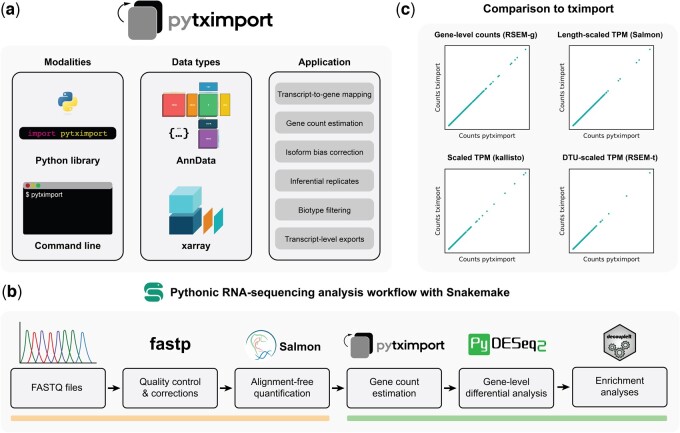
Summary: Transcript quantification tools efficiently map bulk RNA sequencing (RNA-seq) reads to reference transcriptomes. However, their output consists of transcript count estimates that are subject to multiple biases and cannot be readily used with existing differential gene expression analysis tools in Python.Here we present pytximport, a Python implementation of the tximport R package that supports a variety of input formats, different modes of bias correction, inferential replicates, gene-level summarization of transcript counts, transcript-level exports, transcript-to-gene mapping generation, and optional filtering of transcripts by biotype. pytximport is part of the scverse ecosystem of open-source Python software packages for omics analyses and includes both a Python as well as a command-line interface.With pytximport, we propose a bulk RNA-seq analysis workflow based on Bioconda and scverse ecosystem packages, ensuring reproducible analyses through Snakemake rules. We apply this pipeline to a publicly available RNA-seq dataset, demonstrating how pytximport enables the creation of Python-centric workflows capable of providing insights into transcriptomic alterations.
Availability and implementation: pytximport is licensed under the GNU General Public License version 3. The source code is available at https://github.com/complextissue/pytximport and via Zenodo with DOI: 10.5281/zenodo.13907917. A related Snakemake workflow is available through GitHub at https://github.com/complextissue/snakemake-bulk-rna-seq-workflow and Zenodo with DOI: 10.5281/zenodo.12713811. Documentation and a vignette for new users are available at: https://pytximport.readthedocs.io.
© The Author(s) 2024. Published by Oxford University Press.
Figures
Similar articles
-
PyDESeq2: a python package for bulk RNA-seq differential expression analysis.Bioinformatics. 2023 Sep 2;39(9):btad547. doi: 10.1093/bioinformatics/btad547. Bioinformatics. 2023. PMID: 37669147 Free PMC article.
-
RASflow: an RNA-Seq analysis workflow with Snakemake.BMC Bioinformatics. 2020 Mar 18;21(1):110. doi: 10.1186/s12859-020-3433-x. BMC Bioinformatics. 2020. PMID: 32183729 Free PMC article.
-
SPARTA: Simple Program for Automated reference-based bacterial RNA-seq Transcriptome Analysis.BMC Bioinformatics. 2016 Feb 4;17:66. doi: 10.1186/s12859-016-0923-y. BMC Bioinformatics. 2016. PMID: 26847232 Free PMC article.
-
pyrpipe: a Python package for RNA-Seq workflows.NAR Genom Bioinform. 2021 Jun 1;3(2):lqab049. doi: 10.1093/nargab/lqab049. eCollection 2021 Jun. NAR Genom Bioinform. 2021. PMID: 34085037 Free PMC article.
-
Scaling up single-cell RNA-seq data analysis with CellBridge workflow.Bioinformatics. 2023 Dec 1;39(12):btad760. doi: 10.1093/bioinformatics/btad760. Bioinformatics. 2023. PMID: 38113416 Free PMC article.
Cited by
-
Pathology-oriented multiplexing enables integrative disease mapping.Nature. 2025 Aug;644(8076):516-526. doi: 10.1038/s41586-025-09225-2. Epub 2025 Jul 18. Nature. 2025. PMID: 40681898 Free PMC article.
References
-
- He D, Soneson C, Patro R. Understanding and evaluating ambiguity in single-cell and single-nucleus RNA-sequencing. bioRxiv, 10.1101/2023.01.04.522742, 2023, preprint: not peer reviewed. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
