

A temporal cortex cell atlas highlights gene expression dynamics during human brain maturation
- PMID: 39567748
- PMCID: PMC11631765
- DOI: 10.1038/s41588-024-01990-6
A temporal cortex cell atlas highlights gene expression dynamics during human brain maturation
Abstract
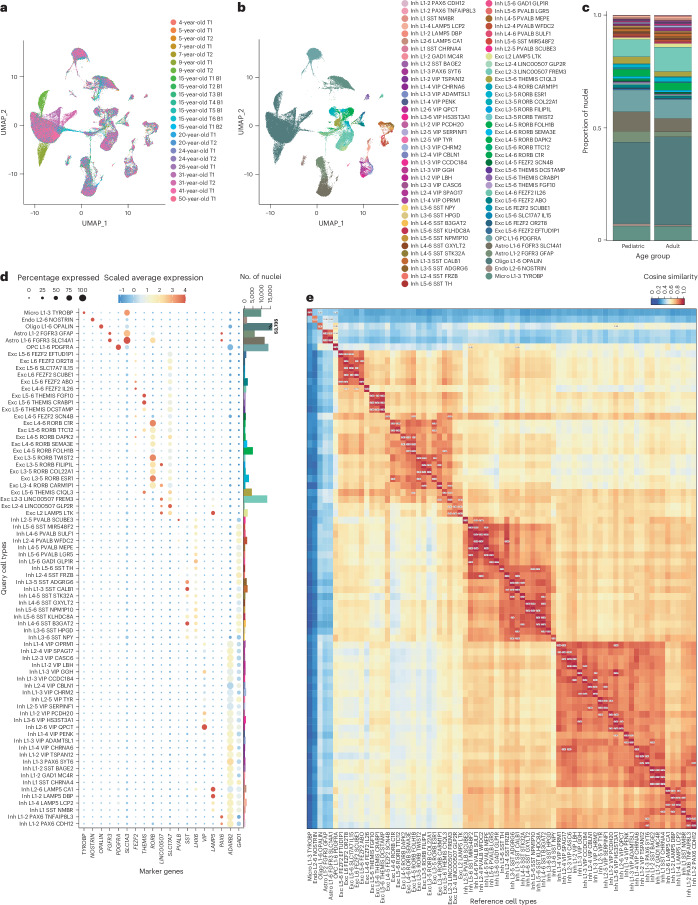
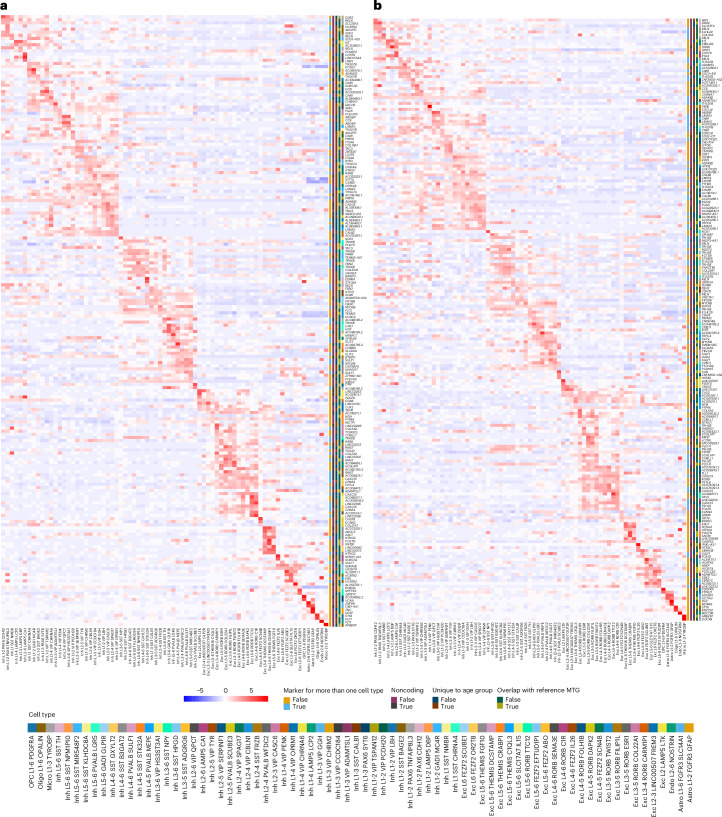
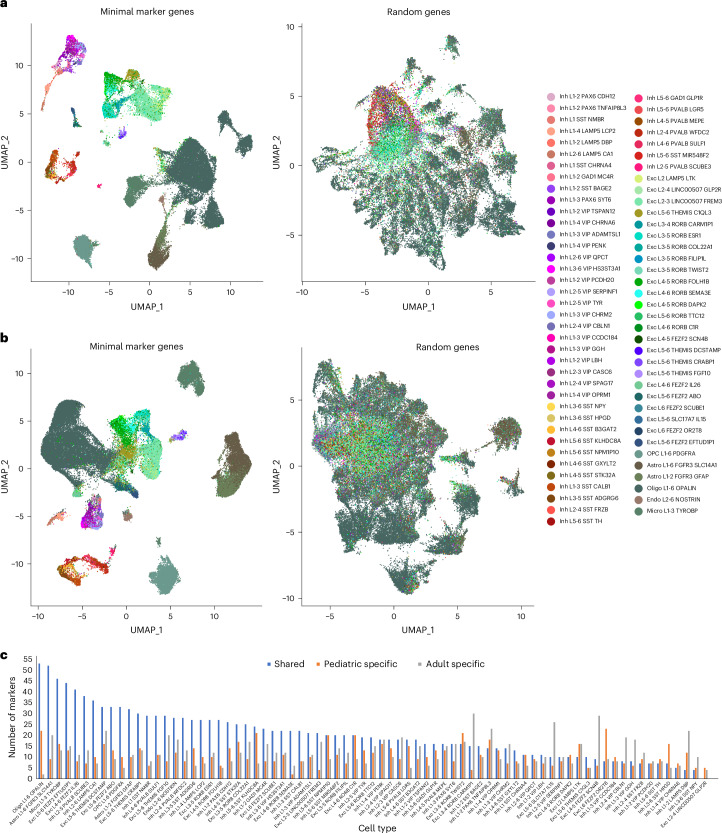
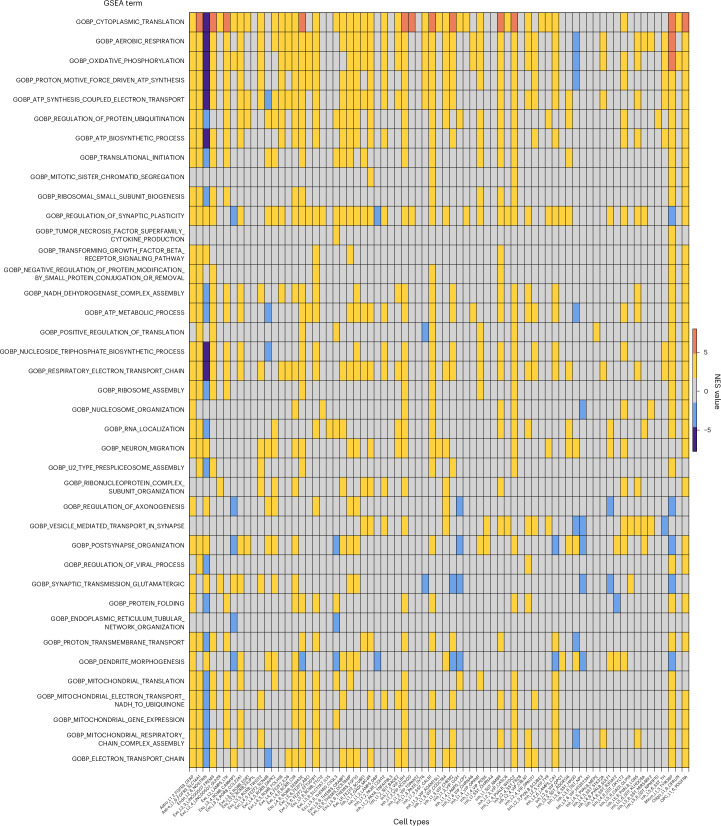
The human brain undergoes protracted postnatal maturation, guided by dynamic changes in gene expression. Most studies exploring these processes have used bulk tissue analyses, which mask cell-type-specific gene expression dynamics. Here, using single-nucleus RNA sequencing on temporal lobe tissue, including samples of African ancestry, we build a joint pediatric and adult atlas of 75 cell subtypes, which we verify with spatial transcriptomics. We explore the differences between pediatric and adult cell subtypes, revealing the genes and pathways that change during brain maturation. Our results highlight excitatory neuron subtypes, including the LTK and FREM subtypes, that show elevated expression of genes associated with cognition and synaptic plasticity in pediatric tissue. The resources we present here improve our understanding of the brain during its development and contribute to global efforts to build an inclusive brain cell map.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures












Update of
-
Cell type-specific gene expression dynamics during human brain maturation.bioRxiv [Preprint]. 2024 May 17:2023.09.29.560114. doi: 10.1101/2023.09.29.560114. bioRxiv. 2024. Update in: Nat Genet. 2024 Dec;56(12):2718-2730. doi: 10.1038/s41588-024-01990-6. PMID: 37808657 Free PMC article. Updated. Preprint.
References
-
- Regev, A. et al. The Human Cell Atlas. eLife10.7554/eLife.27041 (2017).
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
