

Non pathological sweat test, pancreatic insufficiency and Cystic Fibrosis: an unusual case in a child with F508del-duplication of exons 1-3 CFTR genotype
- PMID: 39567905
- PMCID: PMC11577807
- DOI: 10.1186/s12887-024-05154-7
Non pathological sweat test, pancreatic insufficiency and Cystic Fibrosis: an unusual case in a child with F508del-duplication of exons 1-3 CFTR genotype
Abstract
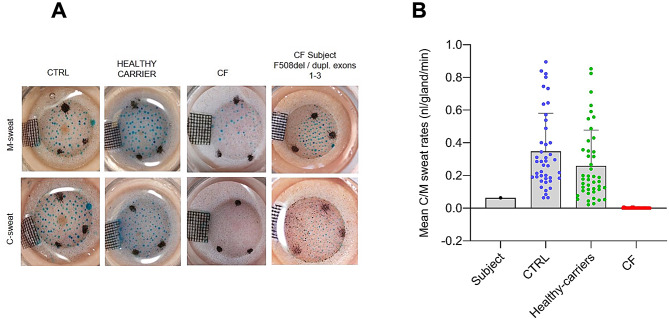
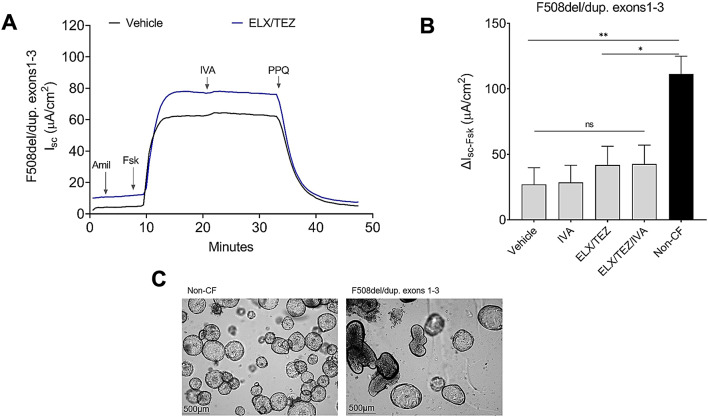
While Cystic Fibrosis is characterized by a high phenotypic variability, a correlation is reported between the pancreatic status and the CFTR genotype. Here we report an unusual case of a child with Cystic Fibrosis (F508del-duplication of exons 1-3 genotype) diagnosed at 8 years old for pancreatic insufficiency and non-pathological sweat test, in absence of respiratory symptoms and acute episodes of pancreatitis. Nasal potential differences and intestinal current measurements were normal, while the short-circuit current measured on patient-derived colonoids grown on Transwell® indicated the presence of a reduced CFTR-dependent current relative to non-CF colonoids with, a modest improvement of CFTR activity record following treatment with elexacaftor/tezacaftor/ivacaftor.This case opens the discussion on the importance of performing CFTR sequencing and the search for large gene rearrangements in cases of pancreatic insufficiency of unclear etiology, also in the presence of non-pathological sweat test. Children with CF and non-pathological sweat chloride are likely to develop higher concentrations if they truly have CF.
Keywords: CFTR; Diagnosis; Duplication; Genotype; Normal; Sweat chloride.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: An approval was obtained from the pediatric ethical committee of the Cystic Fibrosis center in Florence and for the Cystic Fibrosis Center AOUI of Verona (protocol#CFTR050). Consent for publication: Parents provided written informed consent for anonymous data processing and to participate in this case investigation. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del).Cochrane Database Syst Rev. 2020 Dec 17;12(12):CD010966. doi: 10.1002/14651858.CD010966.pub3. Cochrane Database Syst Rev. 2020. Update in: Cochrane Database Syst Rev. 2023 Nov 20;11:CD010966. doi: 10.1002/14651858.CD010966.pub4. PMID: 33331662 Free PMC article. Updated.
-
The expanded French compassionate programme for elexacaftor-tezacaftor-ivacaftor use in people with cystic fibrosis without a F508del CFTR variant: a real-world study.Lancet Respir Med. 2024 Nov;12(11):888-900. doi: 10.1016/S2213-2600(24)00208-X. Epub 2024 Aug 13. Lancet Respir Med. 2024. PMID: 39151434
-
Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial.Lancet. 2019 Nov 23;394(10212):1940-1948. doi: 10.1016/S0140-6736(19)32597-8. Epub 2019 Oct 31. Lancet. 2019. PMID: 31679946 Free PMC article. Clinical Trial.
-
Real-world data confirm elexacftor/tezacaftor/ivacaftor modulators halves sweat chloride concentration in eligible people with cystic fibrosis.APMIS. 2024 Oct;132(10):728-733. doi: 10.1111/apm.13453. Epub 2024 Aug 2. APMIS. 2024. PMID: 39092470
-
Correctors (specific therapies for class II CFTR mutations) for cystic fibrosis.Cochrane Database Syst Rev. 2018 Aug 2;8(8):CD010966. doi: 10.1002/14651858.CD010966.pub2. Cochrane Database Syst Rev. 2018. Update in: Cochrane Database Syst Rev. 2020 Dec 17;12:CD010966. doi: 10.1002/14651858.CD010966.pub3. PMID: 30070364 Free PMC article. Updated. Review.
References
-
- Grasemann H, Ratjen F. Cystic fibrosis. N Engl J Med. 2023;389:1693–707. - PubMed
-
- Terlizzi V, Farrell PM. Update on advances in cystic fibrosis towards a cure and implications for primary care clinicians. Curr Probl Pediatr Adolesc Health Care. 2024;54:101637. - PubMed
-
- Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017; 181S:S4-S15.e1. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
