

Sample multiplexing for retinal single-cell RNA sequencing
- PMID: 39569377
- PMCID: PMC11576387
- DOI: 10.1016/j.isci.2024.111250
Sample multiplexing for retinal single-cell RNA sequencing
Abstract
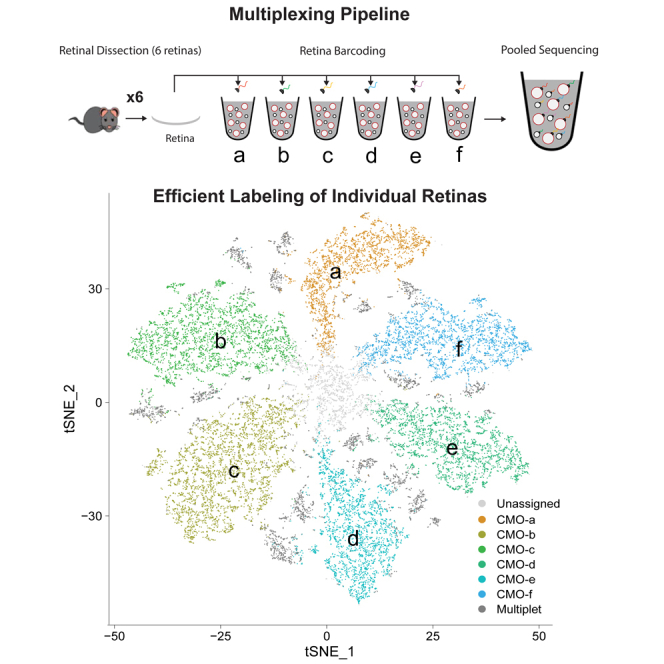
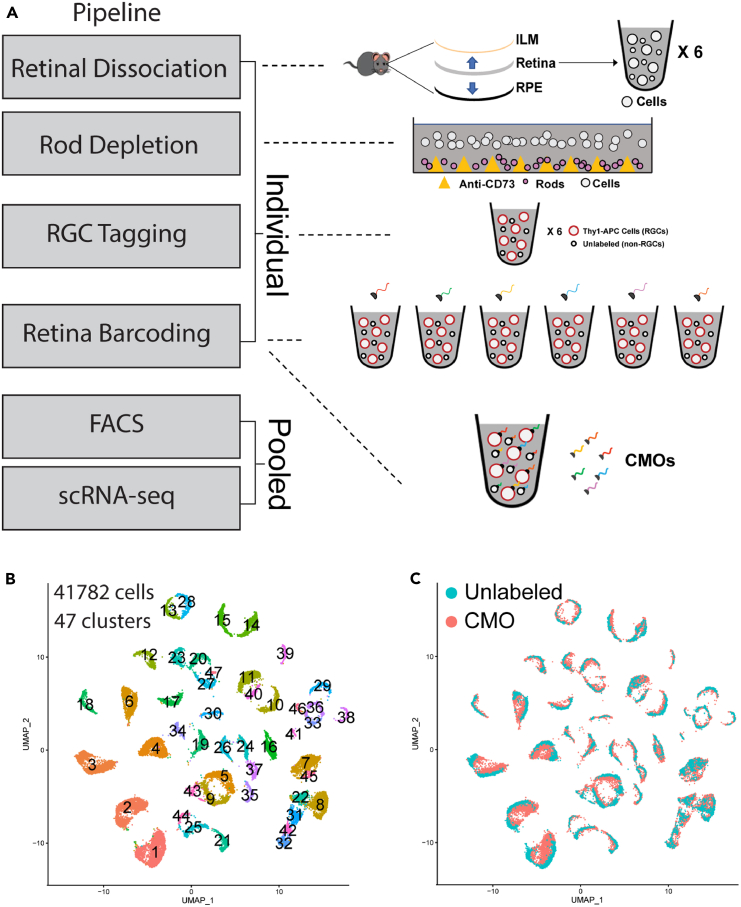
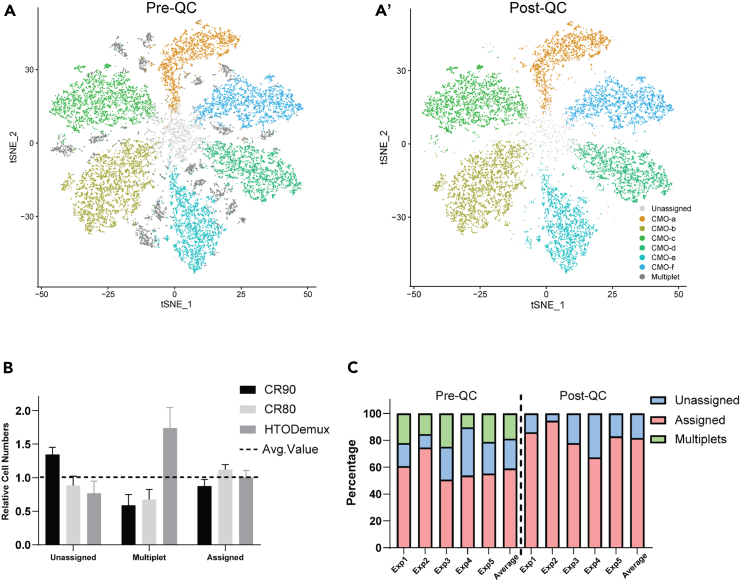
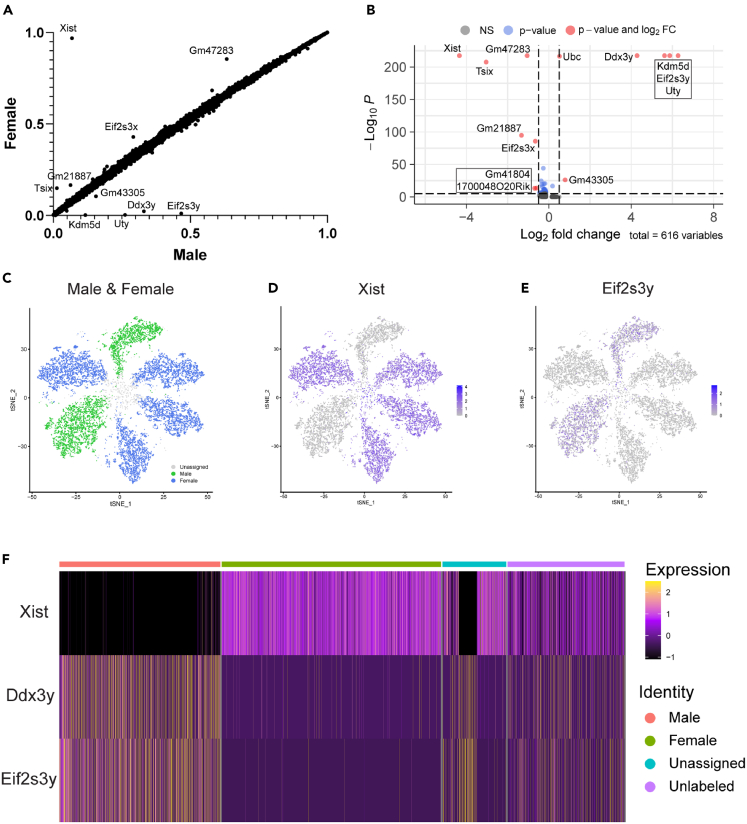
Rare cell populations can be challenging to characterize using microfluidic single-cell RNA sequencing (scRNA-seq) platforms. Typically, the population of interest must be enriched and pooled from multiple biological specimens for efficient collection. However, these practices preclude the resolution of sample origin together with phenotypic data and are problematic in experiments in which biological or technical variation is expected to be high (e.g., disease models, genetic perturbation screens, or human samples). One solution is sample multiplexing whereby each sample is tagged with a unique sequence barcode that is resolved bioinformatically. We have established a scRNA-seq sample multiplexing pipeline for mouse retinal ganglion cells using cholesterol-modified oligos. We utilized the enhanced precision of this dataset to investigate cell type distribution and transcriptomic variance across retinal samples. Additionally, we demonstrate that our multiplexed dataset can be useful for the identification of multiplets in non-labeled samples, a common challenge in scRNA-seq analysis.
Keywords: Molecular biology; Neuroscience; Omics; Transcriptomics.
© 2024 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Sample multiplexing for retinal single-cell RNA-sequencing.bioRxiv [Preprint]. 2024 Apr 26:2024.04.23.589797. doi: 10.1101/2024.04.23.589797. bioRxiv. 2024. Update in: iScience. 2024 Oct 24;27(11):111250. doi: 10.1016/j.isci.2024.111250. PMID: 38712294 Free PMC article. Updated. Preprint.
References
-
- Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
