

Phenotype-genotype correlation in X-linked Charcot-Marie-Tooth disease: A French cohort study
- PMID: 39569692
- PMCID: PMC11622270
- DOI: 10.1111/ene.16523
Phenotype-genotype correlation in X-linked Charcot-Marie-Tooth disease: A French cohort study
Abstract
Background and purpose: X-linked Charcot-Marie-Tooth disease type 1 (CMTX1) ranks as the second most prevalent hereditary neuropathy and, currently, has no definitive cure. Emerging preclinical trials offer hope for potential clinical studies in the near future. While it is widely accepted that experimental groups in these trials should be balanced for age and gender, there is a current shortfall in data regarding phenotype-genotype correlations. Our aim was to provide a more detailed understanding of these correlations to facilitate the formation of well-matched patient groups in upcoming clinical trials.
Methods: We conducted a retrospective evaluation of CMTX1 patients from 13 designated reference centers in France. Data on genetics, clinical features, and nerve conduction were systematically gathered.
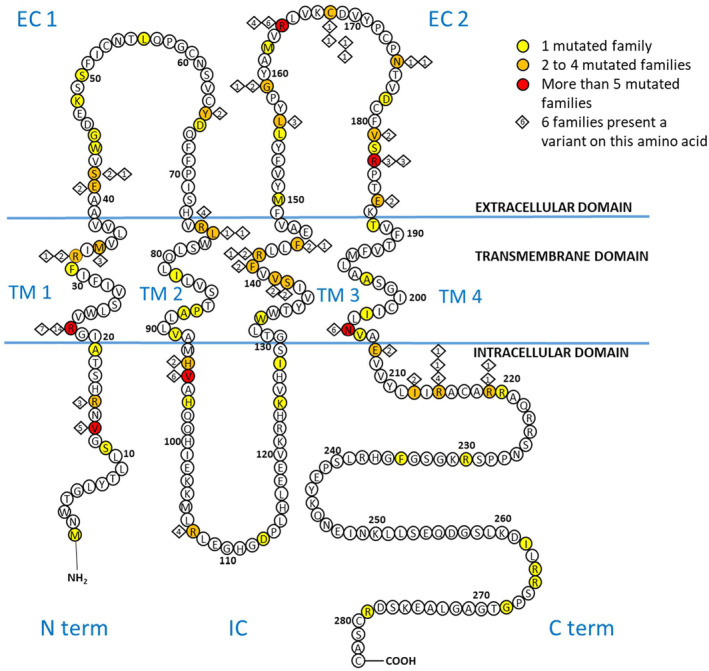
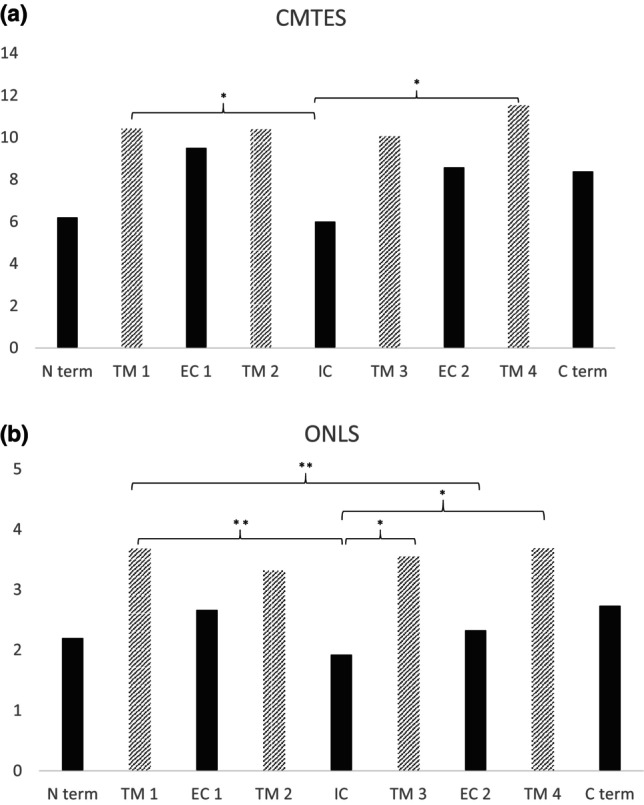
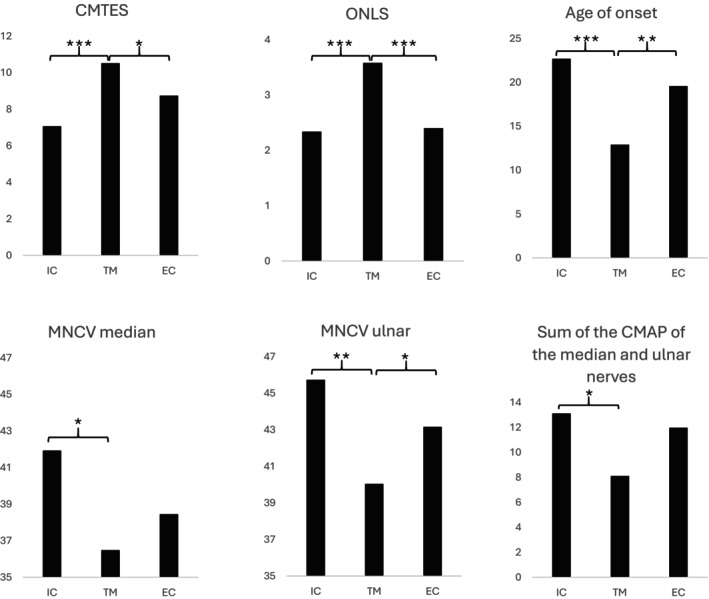
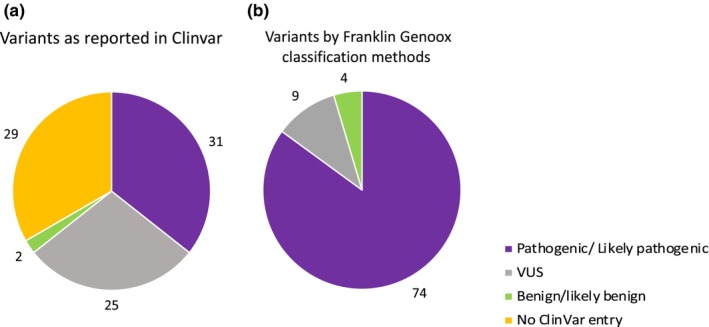
Results: We analyzed the genotype-phenotype correlations in 275 CMTX1 patients belonging to 162 families and carrying 87 distinct variants. Patients with variants affecting the transmembrane domains demonstrated significantly greater severity, as evidenced by a Charcot-Marie-Tooth Examination Score of 10.5, compared to 7.1 for those with intracellular domain variants and 8.7 for extracellular domain variants (p < 0.000). These patients also experienced an earlier age of onset, showed slower ulnar nerve conduction velocities and had more substantial loss of motor amplitude.
Conclusions: This study confirms the presence of a correlation between the mutated protein domain and the clinical phenotype. Patients with a variant in the transmembrane domains demonstrated a more severe clinical and electrophysiological profile. Consequently, the genotype could play a prognostic role in addition to its diagnostic role, and it will be essential to consider this in future clinical trials.
Keywords: CMTX; Charcot‐Marie‐Tooth; clinical trial; connexine 32; hereditary peripheral neuropathy.
© 2024 The Author(s). European Journal of Neurology published by John Wiley & Sons Ltd on behalf of European Academy of Neurology.
Conflict of interest statement
The authors report no disclosures relevant to the article.
Figures
References
-
- Bergoffen J, Scherer SS, Wang S, et al. Connexin mutations in X‐linked Charcot–Marie–tooth disease. Science. 1993;262(5142):2039‐2042. - PubMed
-
- La BN. Maladie de Charcot–Marie‐tooth. Presse Médicale févr. 2009;38(2):200‐209. - PubMed
-
- Hahn AF, Bolton CF, White CM, et al. Genotype/phenotype correlations in X‐linked dominant Charcot–Marie–tooth disease. Ann N Y Acad Sci. 1999;883:366‐382. - PubMed
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
