

ZBTB7A is a modulator of KDM5-driven transcriptional networks in basal breast cancer
- PMID: 39570746
- PMCID: PMC11694571
- DOI: 10.1016/j.celrep.2024.114991
ZBTB7A is a modulator of KDM5-driven transcriptional networks in basal breast cancer
Abstract
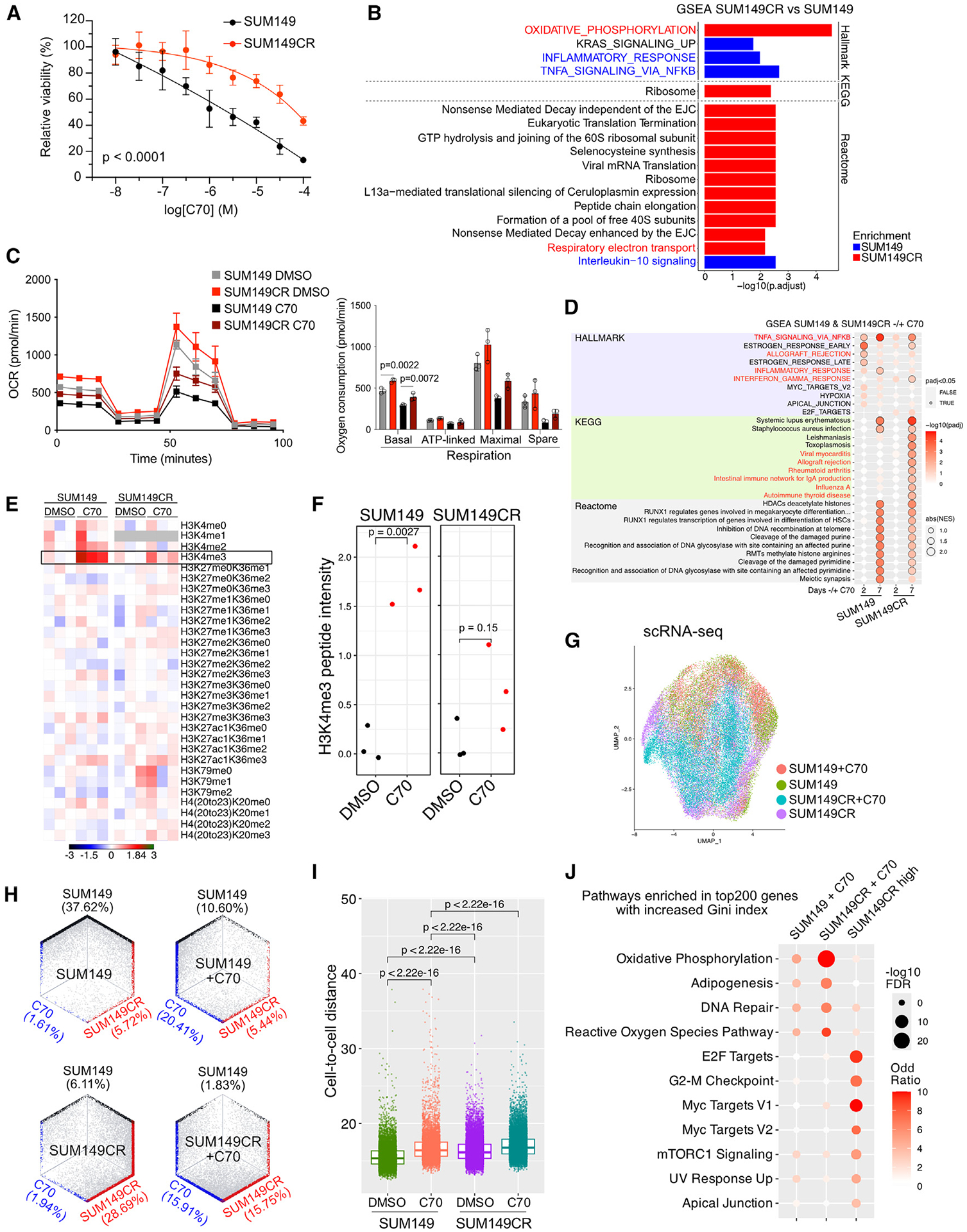
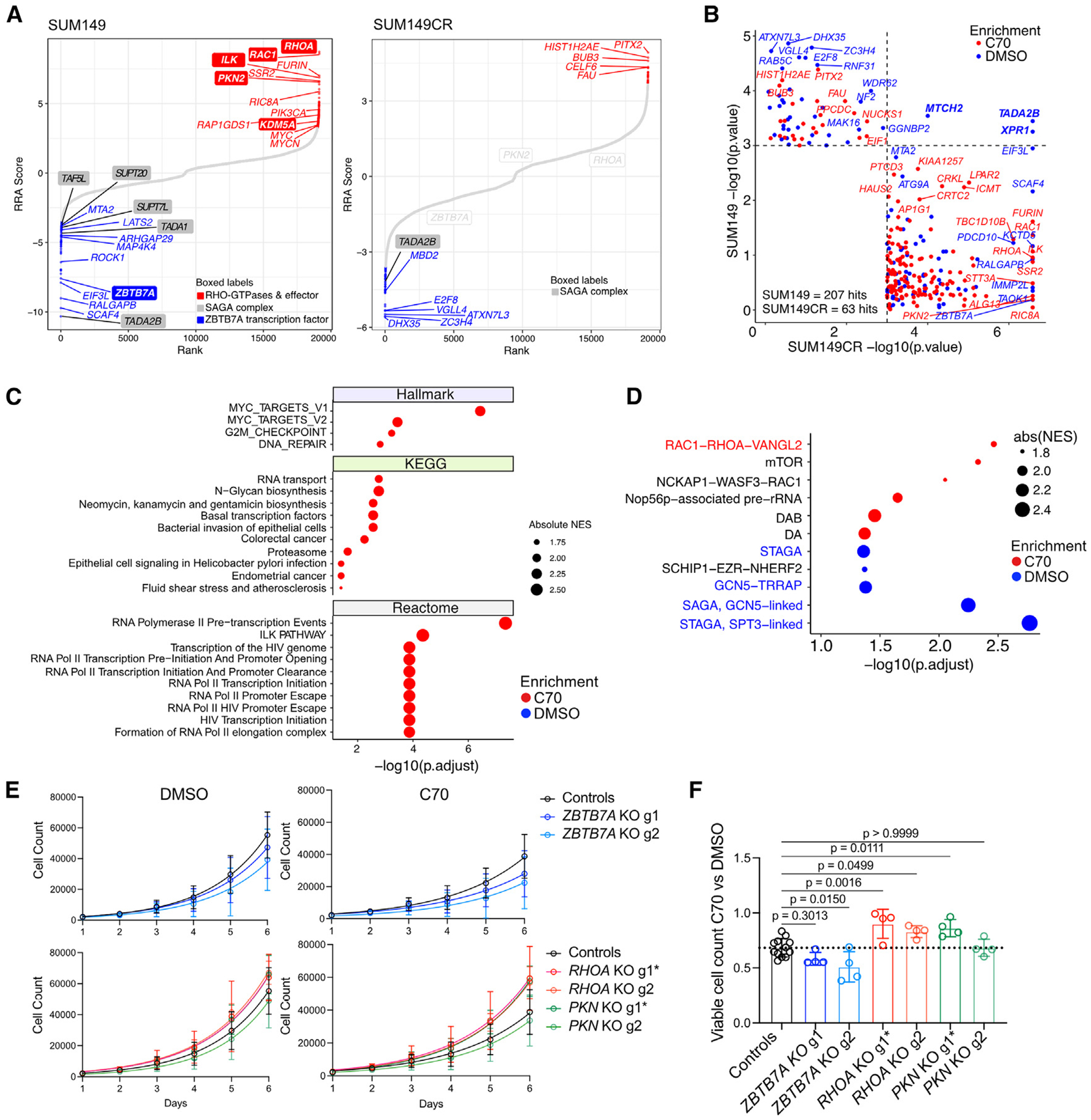
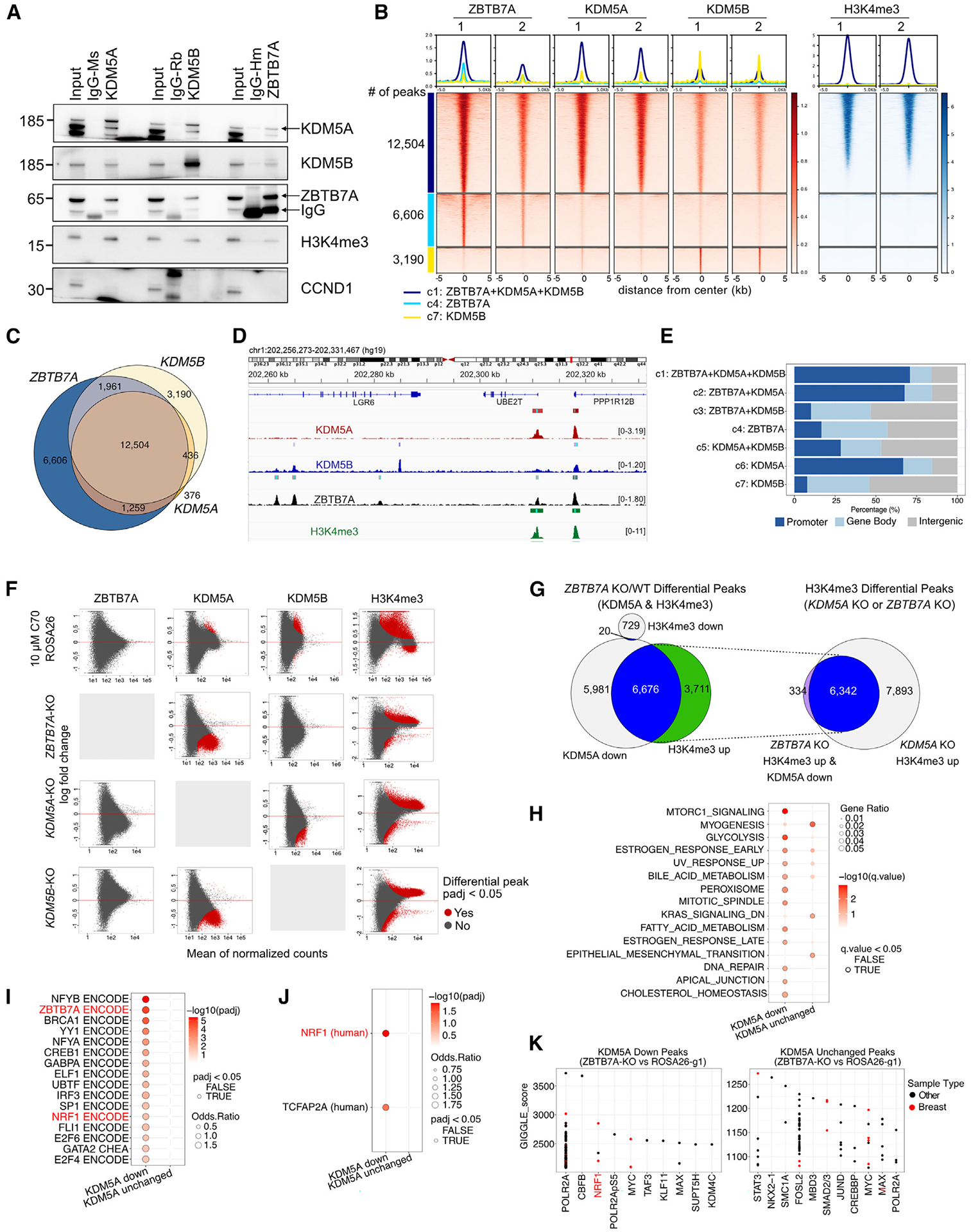
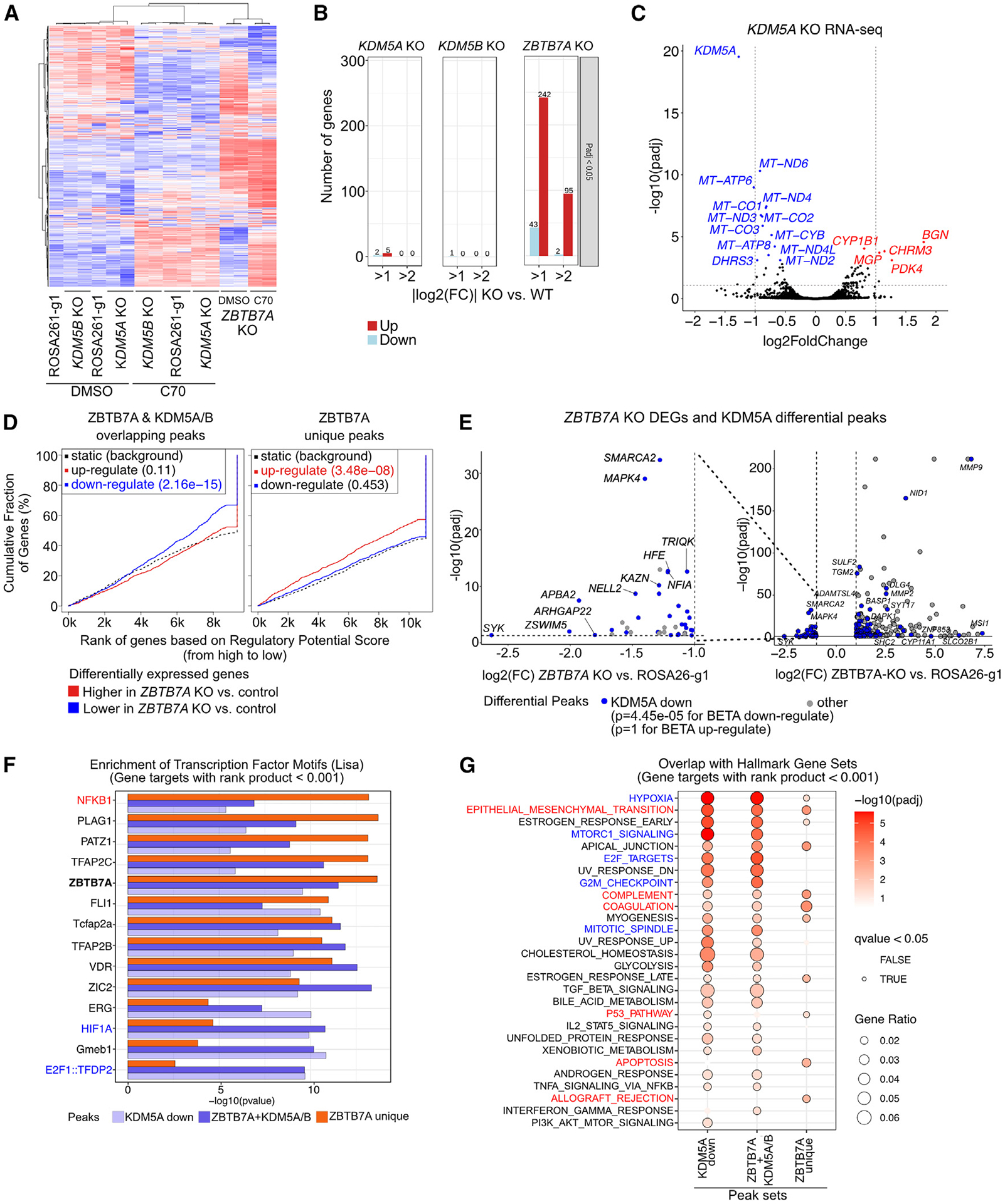
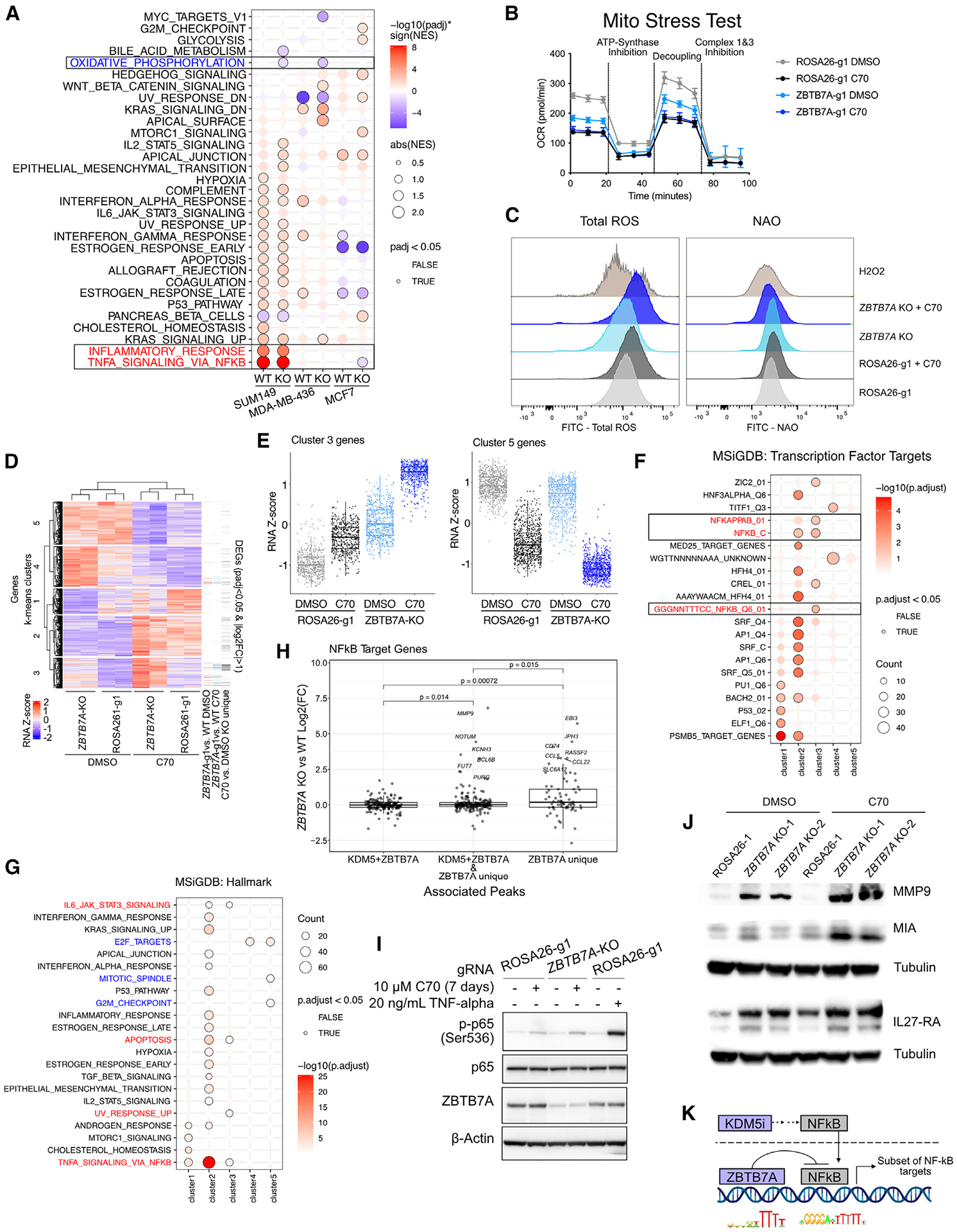
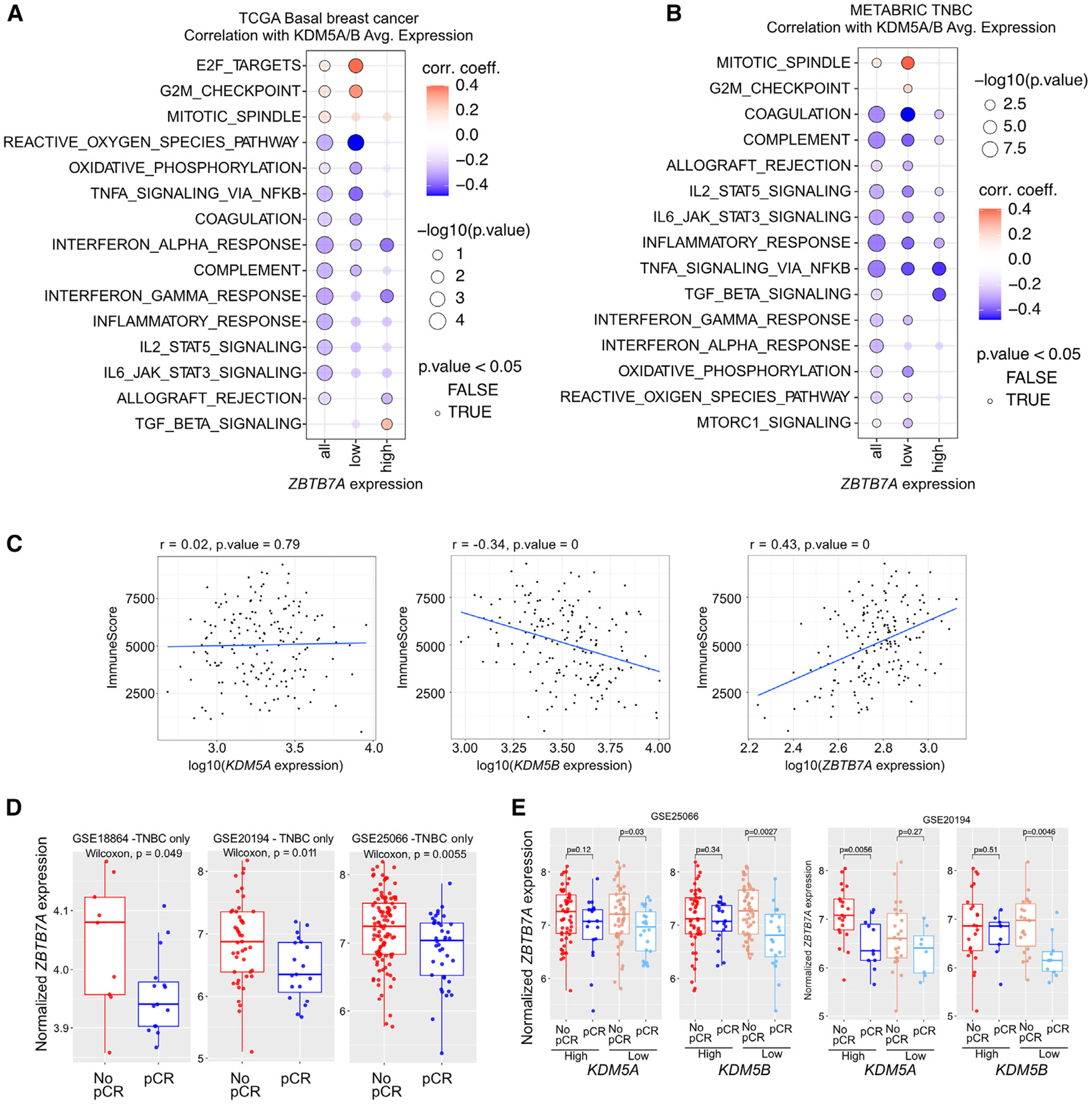
We previously described that the KDM5B histone H3 lysine 4 demethylase is an oncogene in estrogen-receptor-positive breast cancer. Here, we report that KDM5A is amplified and overexpressed in basal breast tumors, and KDM5 inhibition (KDM5i) suppresses the growth of KDM5-amplified breast cancer cell lines. Using CRISPR knockout screens in a basal breast cancer cell line with or without KDM5i, we found that deletion of the ZBTB7A transcription factor and core SAGA complex sensitizes cells to KDM5i, whereas deletion of RHO-GTPases leads to resistance. Chromatin immunoprecipitation sequencing (ChIP-seq) and RNA sequencing (RNA-seq) revealed co-localization of ZBTB7A and KDM5A/B at promoters with high histone H3K4me3 and dependence of KDM5A chromatin binding on ZBTB7A. ZBTB7A knockout altered the transcriptional response to KDM5i at NF-κB targets and mitochondrion-related pathways. High expression of ZBTB7A in triple-negative breast cancer is significantly associated with poor response to neoadjuvant chemotherapy. Our work furthers the understanding of KDM5-mediated gene regulation and identifies mediators of sensitivity to KDM5i.
Keywords: CP: Cancer; CRISPR screen; breast cancer; histone demethylase; mitochondrial signaling.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.P. serves on the scientific advisory boards of Ideaya Biosciences and Scorpion Therapeutics, holds equity options in Scorpion Therapeutics and Ideaya Biosciences, and receives sponsored research funding from Novartis, where she also consults. H.W.L. receives research funding from Novartis.
Figures
References
-
- Yuan S, Norgard RJ, and Stanger BZ (2019). Cellular Plasticity in Cancer. Cancer Discov. 9, 837–851. 10.1158/2159-8290.CD-19-0015. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
