

Multimodal single cell-resolved spatial proteomics reveal pancreatic tumor heterogeneity
- PMID: 39572534
- PMCID: PMC11582669
- DOI: 10.1038/s41467-024-54438-0
Multimodal single cell-resolved spatial proteomics reveal pancreatic tumor heterogeneity
Abstract
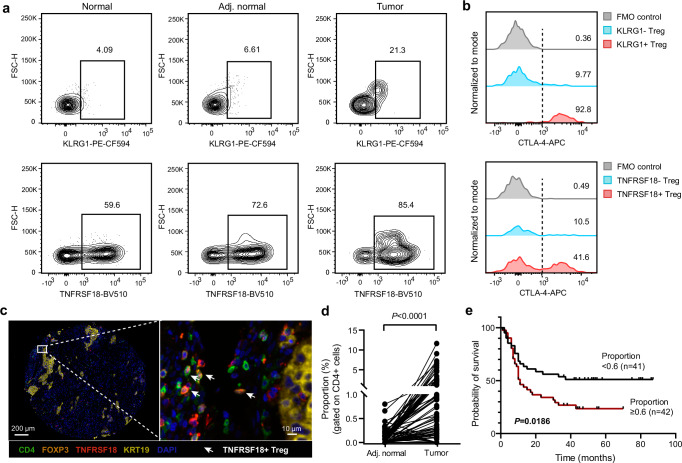
Despite the advances in antibody-guided cell typing and mass spectrometry-based proteomics, their integration is hindered by challenges for processing rare cells in the heterogeneous tissue context. Here, we introduce Spatial and Cell-type Proteomics (SCPro), which combines multiplexed imaging and flow cytometry with ion exchange-based protein aggregation capture technology to characterize spatial proteome heterogeneity with single-cell resolution. The SCPro is employed to explore the pancreatic tumor microenvironment and reveals the spatial alternations of over 5000 proteins by automatically dissecting up to 100 single cells guided by multi-color imaging of centimeter-scale formalin-fixed, paraffin-embedded tissue slide. To enhance cell-type resolution, we characterize the proteome of 14 different cell types by sorting up to 1000 cells from the same tumor, which allows us to deconvolute the spatial distribution of immune cell subtypes and leads to the discovery of subtypes of regulatory T cells. Together, the SCPro provides a multimodal spatial proteomics approach for profiling tissue proteome heterogeneity.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: R.T. is the founder of BayOmics, Inc. The other authors declare no competing interests.
Figures






References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
