

Multi-layered proteomics identifies insulin-induced upregulation of the EphA2 receptor via the ERK pathway which is dependent on low IGF1R level
- PMID: 39572596
- PMCID: PMC11582730
- DOI: 10.1038/s41598-024-77817-5
Multi-layered proteomics identifies insulin-induced upregulation of the EphA2 receptor via the ERK pathway which is dependent on low IGF1R level
Abstract
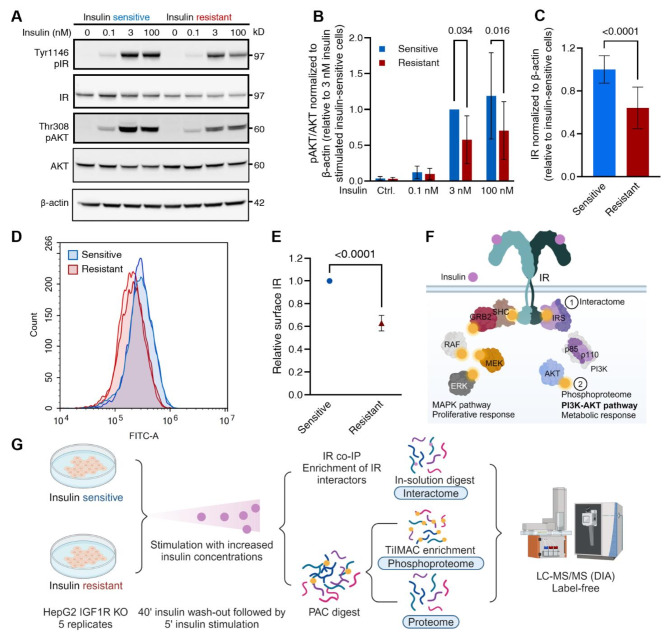
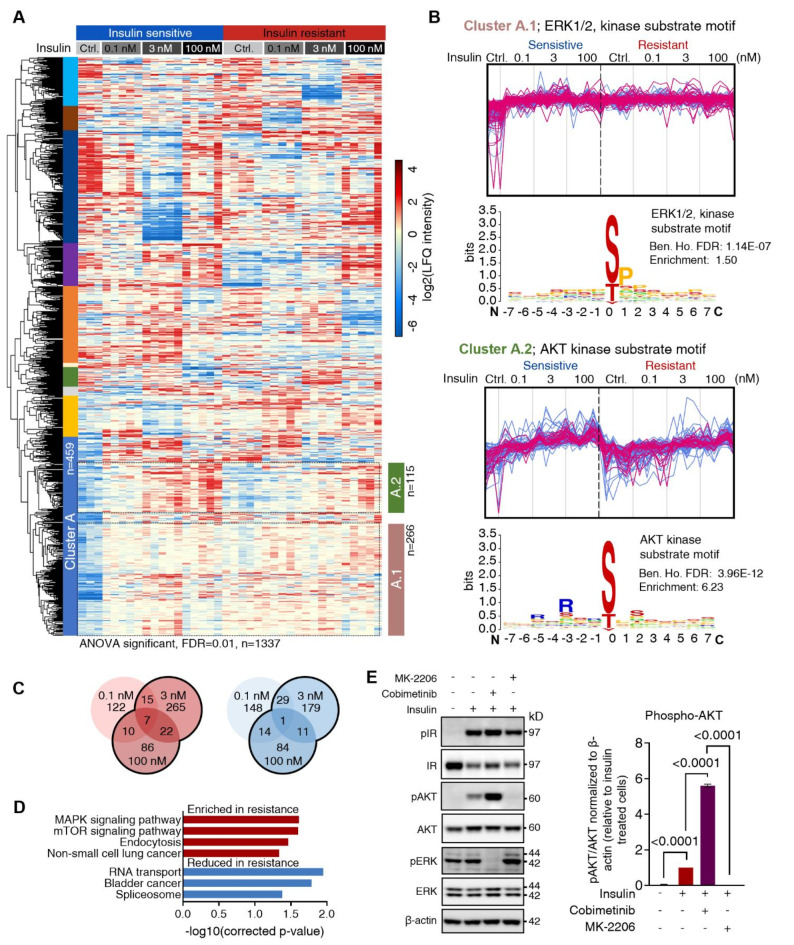
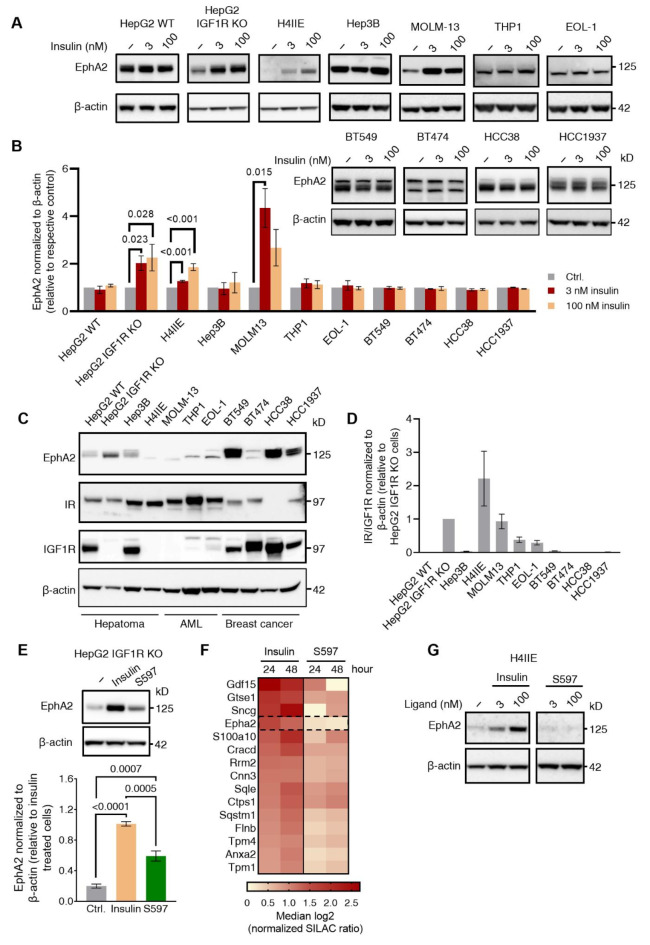
Insulin resistance impairs the cellular insulin response, and often precedes metabolic disorders, like type 2 diabetes, impacting an increasing number of people globally. Understanding the molecular mechanisms in hepatic insulin resistance is essential for early preventive treatments. To elucidate changes in insulin signal transduction associated with hepatocellular resistance, we employed a multi-layered mass spectrometry-based proteomics approach focused on insulin receptor (IR) signaling at the interactome, phosphoproteome, and proteome levels in a long-term hyperinsulinemia-induced insulin-resistant HepG2 cell line with a knockout of the insulin-like growth factor 1 receptor (IGF1R KO). The analysis revealed insulin-stimulated recruitment of the PI3K complex in both insulin-sensitive and -resistant cells. Phosphoproteomics showed attenuated signaling via the metabolic PI3K-AKT pathway but sustained extracellular signal-regulated kinase (ERK) activity in insulin-resistant cells. At the proteome level, the ephrin type-A receptor 2 (EphA2) showed an insulin-induced increase in expression, which occurred through the ERK signaling pathway and was concordantly independent of insulin resistance. Induction of EphA2 by insulin was confirmed in additional cell lines and observed uniquely in cells with high IR-to-IGF1R ratio. The multi-layered proteomics dataset provided insights into insulin signaling, serving as a resource to generate and test hypotheses, leading to an improved understanding of insulin resistance.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Competing interests: S.H.J, A.K.P, H.F.K, D.D, P.K.N, R.S, L.N.A, T.Å.P, and M.G are current or previous employees at Novo Nordisk A/S and are shareholders in the company. CPR is supported by a grant from the Novo Nordisk Foundation. K.B.E and J.V.O declare no competing interests.
Figures



References
-
- Freeman, A. M., Pennings, N. & Acevedo, L. A. Insulin Resistance (StatPearls Publishing, 2023). - PubMed
-
- Johnson, A. M. F. & Olefsky, J. M. The Origins and Drivers of Insulin Resistance. 10.1016/j.cell.2013.01.041 (Elsevier B.V., 2013). - PubMed
-
- Nolan, C. J. & Prentki, M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: Time for a conceptual framework shift. Diab. Vasc. Dis. Res.16(2), 118–127. 10.1177/1479164119827611 (2019). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
