

Exome sequencing in Asian populations identifies low-frequency and rare coding variation influencing Parkinson's disease risk
- PMID: 39572736
- PMCID: PMC11839463
- DOI: 10.1038/s43587-024-00760-7
Exome sequencing in Asian populations identifies low-frequency and rare coding variation influencing Parkinson's disease risk
Abstract
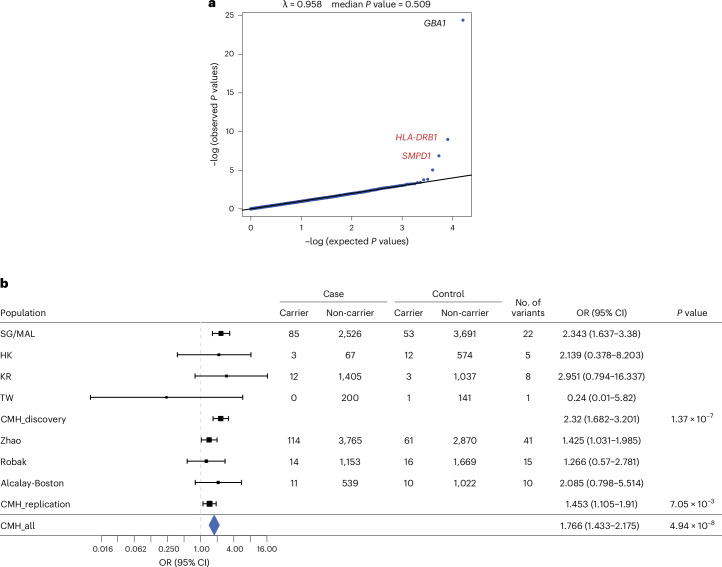
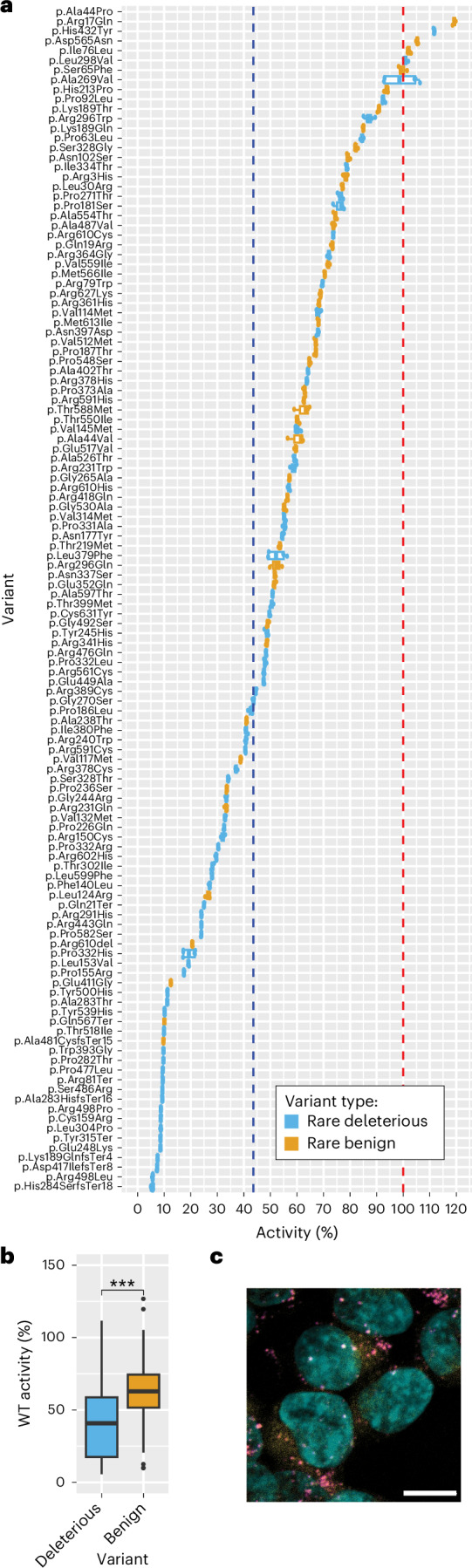
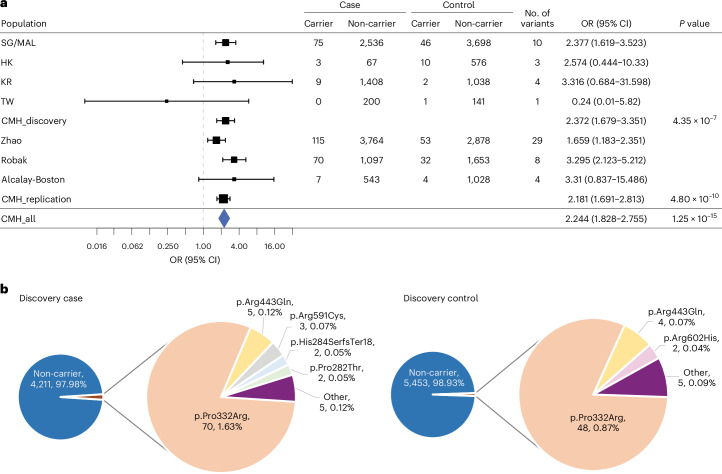
Parkinson's disease (PD) is an incurable, progressive and common movement disorder that is increasing in incidence globally because of population aging. We hypothesized that the landscape of rare, protein-altering variants could provide further insights into disease pathogenesis. Here we performed whole-exome sequencing followed by gene-based tests on 4,298 PD cases and 5,512 controls of Asian ancestry. We showed that GBA1 and SMPD1 were significantly associated with PD risk, with replication in a further 5,585 PD cases and 5,642 controls. We further refined variant classification using in vitro assays and showed that SMPD1 variants with reduced enzymatic activity display the strongest association (<44% activity, odds ratio (OR) = 2.24, P = 1.25 × 10-15) with PD risk. Moreover, 80.5% of SMPD1 carriers harbored the Asian-specific p.Pro332Arg variant (OR = 2.16; P = 4.47 × 10-8). Our findings highlight the utility of performing exome sequencing in diverse ancestry groups to identify rare protein-altering variants in genes previously unassociated with disease.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: Z.G.-O. received consultancy fees from Lysosomal Therapeutics, Idorsia, Prevail Therapeutics, Ono Therapeutics, Denali, Handl Therapeutics, Neuron23, Bial Biotech, Bial, UCB, Capsida, Vanqua Bio, Congruence Therapeutics, Takeda, Jazz Pharmaceuticals, Guidepoint, Lighthouse and Deerfield. R.N.A. received consultation fees from Biogen, Biohaven, Capsida, Gain Therapeutics, Genzyme/Sanofi, Janssen, Servier, SK Biopharmaceuticals, Takeda and Vanqua Bio. Y.X. holds a stock option in NeoCytogen Therapeutics where she is scientific co-founder and Chief Scientific Officer. M.A.N.’s participation in this project was part of a competitive contract awarded to DataTecnica, LLC by the National Institutes of Health to support open science research. M.A.N. also owns stock in Character Bio, Inc. and Neuron23, Inc. The other authors declare no competing interests.
Figures








References
-
- Nutt, J. G. & Wooten, G. F. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N. Engl. J. Med.353, 1021–1027 (2005). - PubMed
-
- Bloem, B. R., Okun, M. S. & Klein, C. Parkinson’s disease. Lancet397, 2284–2303 (2021). - PubMed
-
- Satake, W. et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet.41, 1303–1307 (2009). - PubMed
MeSH terms
Substances
Grants and funding
- MOE-T2EP30220-0008/Ministry of Education - Singapore (MOE)
- MOH-000435/MOH | National Medical Research Council (NMRC)
- MOH-001110/MOH | National Medical Research Council (NMRC)
- OT2 OD032100/OD/NIH HHS/United States
- OT2 OD027060/OD/NIH HHS/United States
- MOH-000207/MOH | National Medical Research Council (NMRC)
- R01 EY015473/EY/NEI NIH HHS/United States
- MOH-001329/Ministry of Health -Singapore (MOH)
- MOH-001110/Ministry of Health -Singapore (MOH)
- MOE-MOET32020-0004/Ministry of Education - Singapore (MOE)
- MOH-001072/MOH | National Medical Research Council (NMRC)
- MOH-000559/MOH | National Medical Research Council (NMRC)
- OT2 OD027852/OD/NIH HHS/United States
- MOE-T2EP30220-0005/Ministry of Education - Singapore (MOE)
- P30 EY014104/EY/NEI NIH HHS/United States
- MOH-001214/MOH | National Medical Research Council (NMRC)
LinkOut - more resources
Full Text Sources
Medical
