

Effects of virus-induced immunogenic cues on oncolytic virotherapy
- PMID: 39572761
- PMCID: PMC11582614
- DOI: 10.1038/s41598-024-80542-8
Effects of virus-induced immunogenic cues on oncolytic virotherapy
Abstract
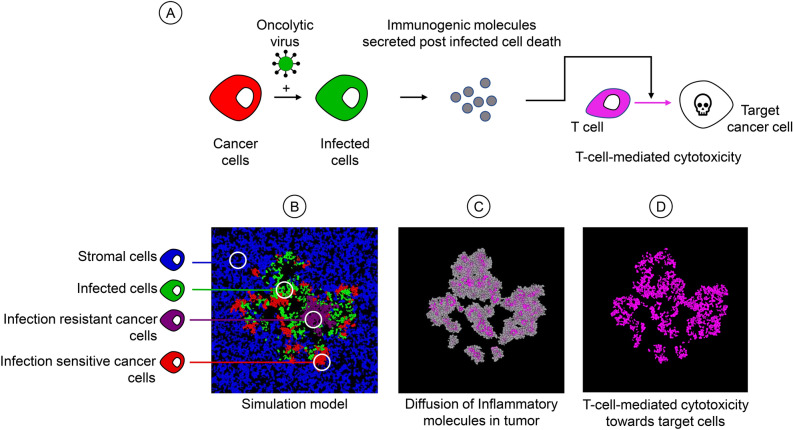
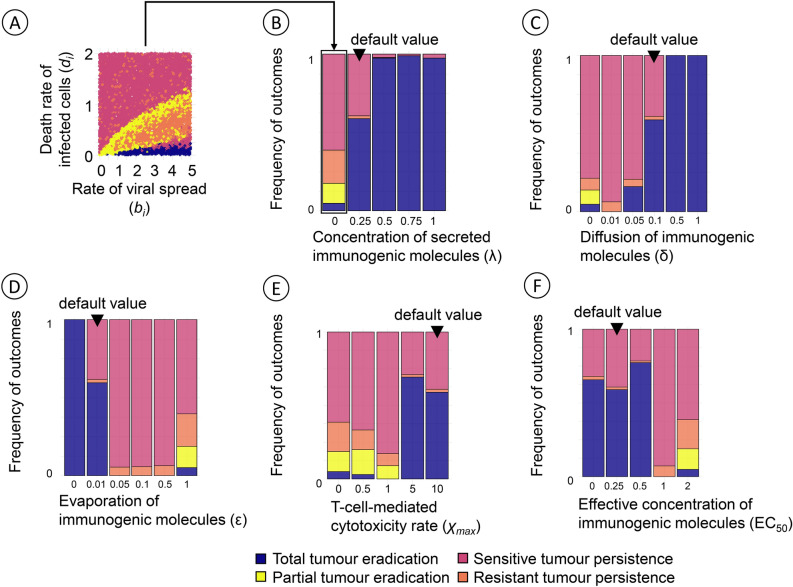
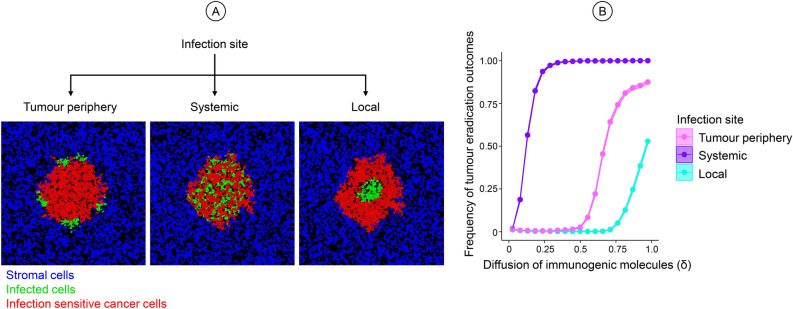
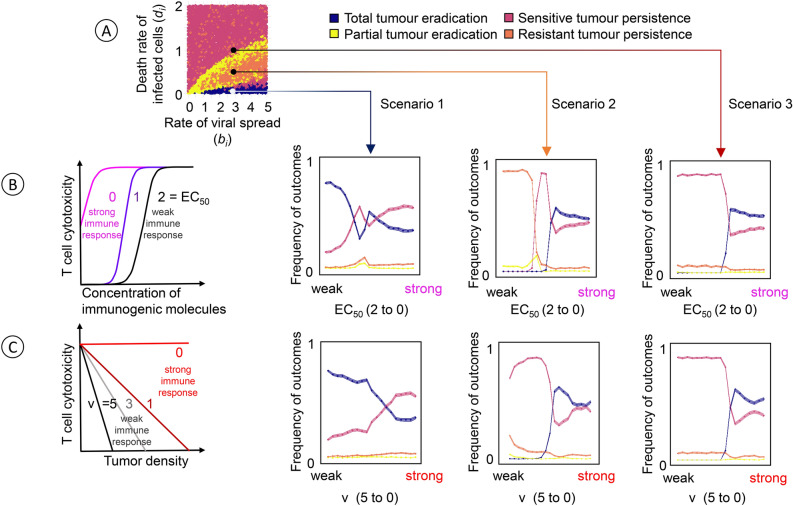
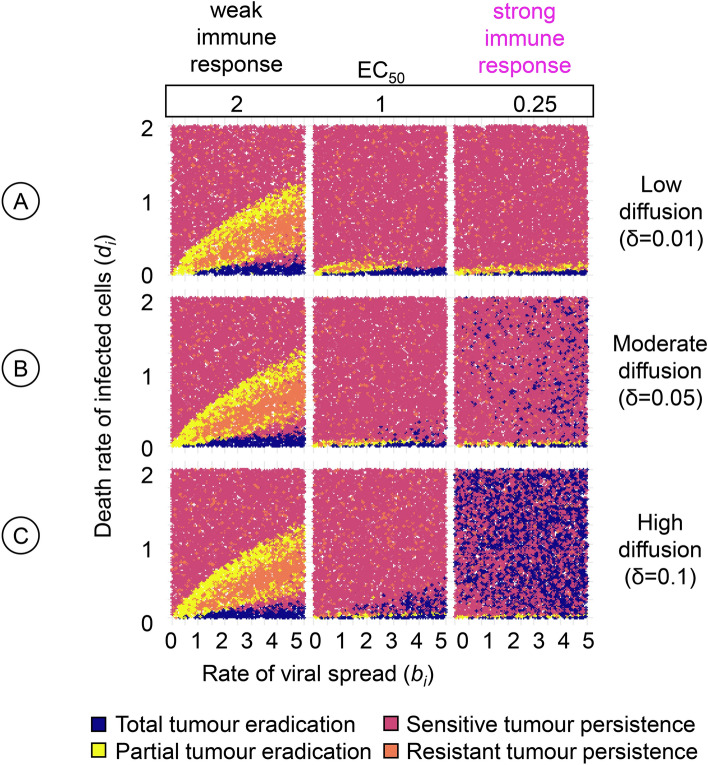
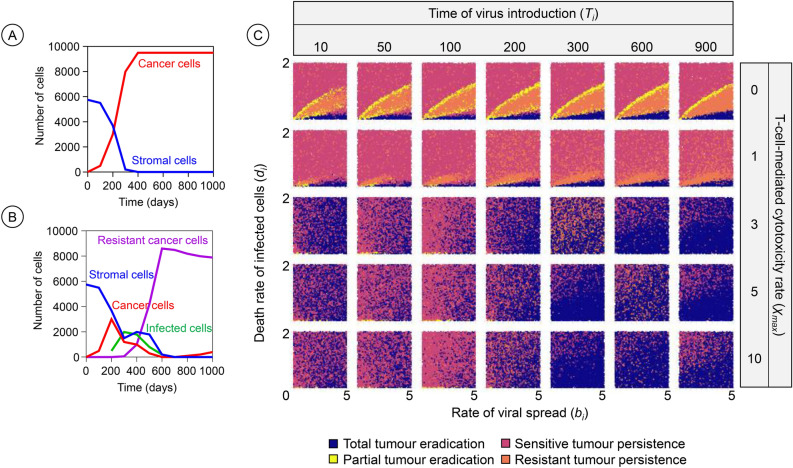
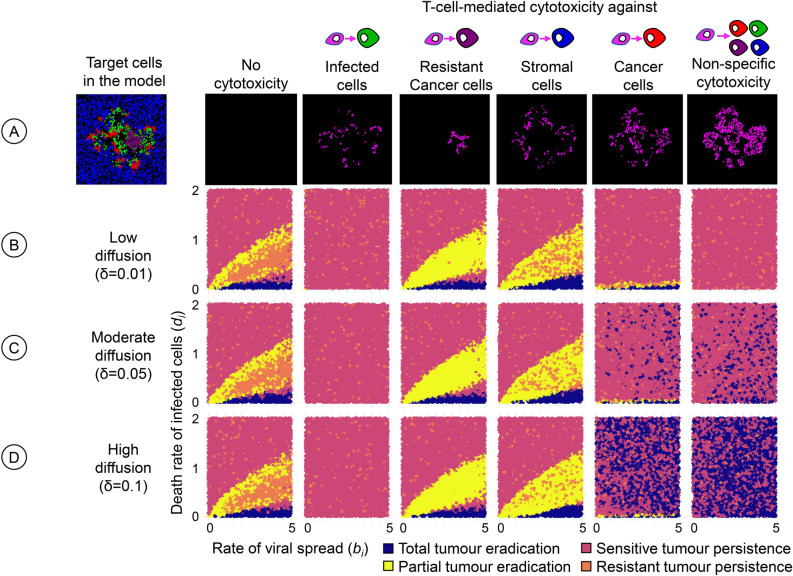
Oncolytic virotherapy is a promising form of cancer treatment that uses viruses to infect and kill cancer cells. In addition to their direct effects on cancer cells, the viruses stimulate various immune responses partly directed against the tumour. Efforts are made to genetically engineer oncolytic viruses to enhance their immunogenic potential. However, the interplay between tumour growth, viral infection, and immune responses is complex and not fully understood, leading to variable and sometimes counterintuitive therapeutic outcomes. Here, we employ a spatio-temporal model to shed more light on this interplay. We investigate systematically how the properties of virus-induced immunogenic signals (their half-life, rate of spread, and potential to promote T-cell-mediated cytotoxicity) affect the therapeutic outcome. Our simulations reveal that strong immunogenic signals, combined with faster diffusion rates, improve the spread of immune activation, leading to better tumour eradication. However, replicate simulations suggest that the outcome of virotherapy is more stochastic than generally appreciated. Our model shows that virus-induced immune responses can interfere with virotherapy, by targeting virus-infected cancer cells and/or by impeding viral spread. In the presence of immune responses, the mode of virus introduction is important, with systemic viral delivery throughout the tumour yielding the most favourable outcomes. The timing of virus introduction also plays a critical role; depending on the efficacy of the immune response, a later start of virotherapy can be advantageous. Overall, our results emphasise that the rational design of oncolytic viruses requires optimising virus-induced immunogenic signals and strategies that balance viral spread with immune activity for improved therapeutic success.
Keywords: Immune response; Immunogenic molecules; Spatial model; Stochastic outcome; Therapy failure.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
