

Genome-wide association analysis provides insights into the molecular etiology of dilated cardiomyopathy
- PMID: 39572783
- PMCID: PMC11631752
- DOI: 10.1038/s41588-024-01952-y
Genome-wide association analysis provides insights into the molecular etiology of dilated cardiomyopathy
Abstract
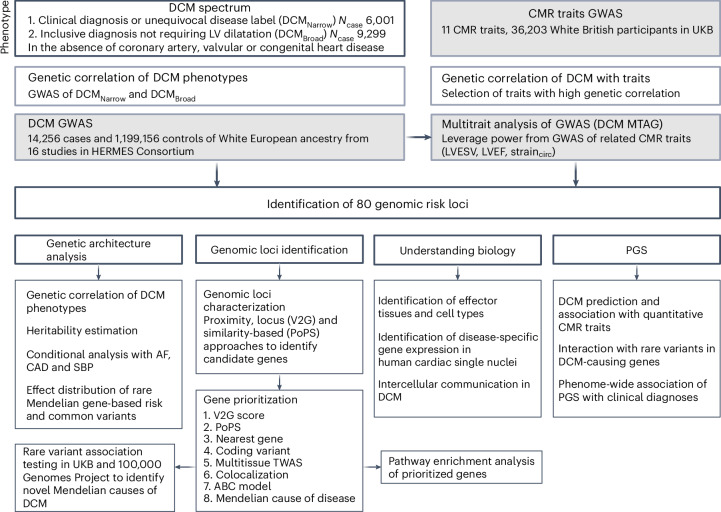
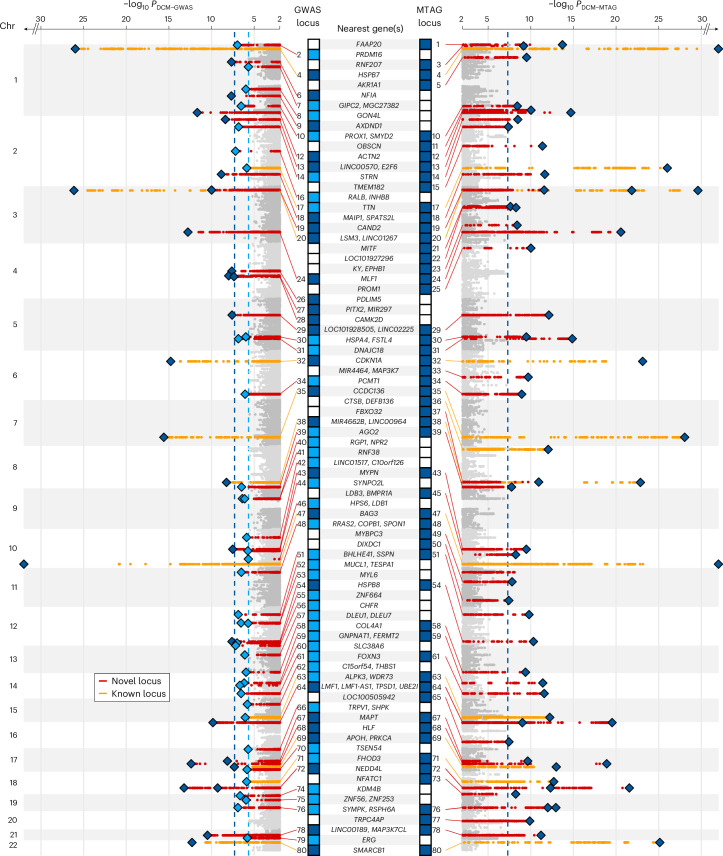
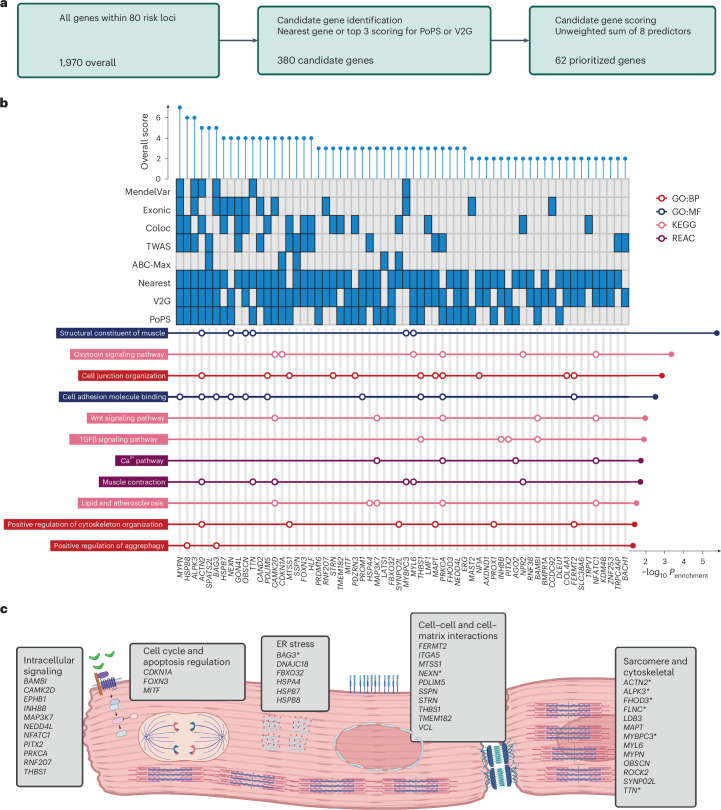
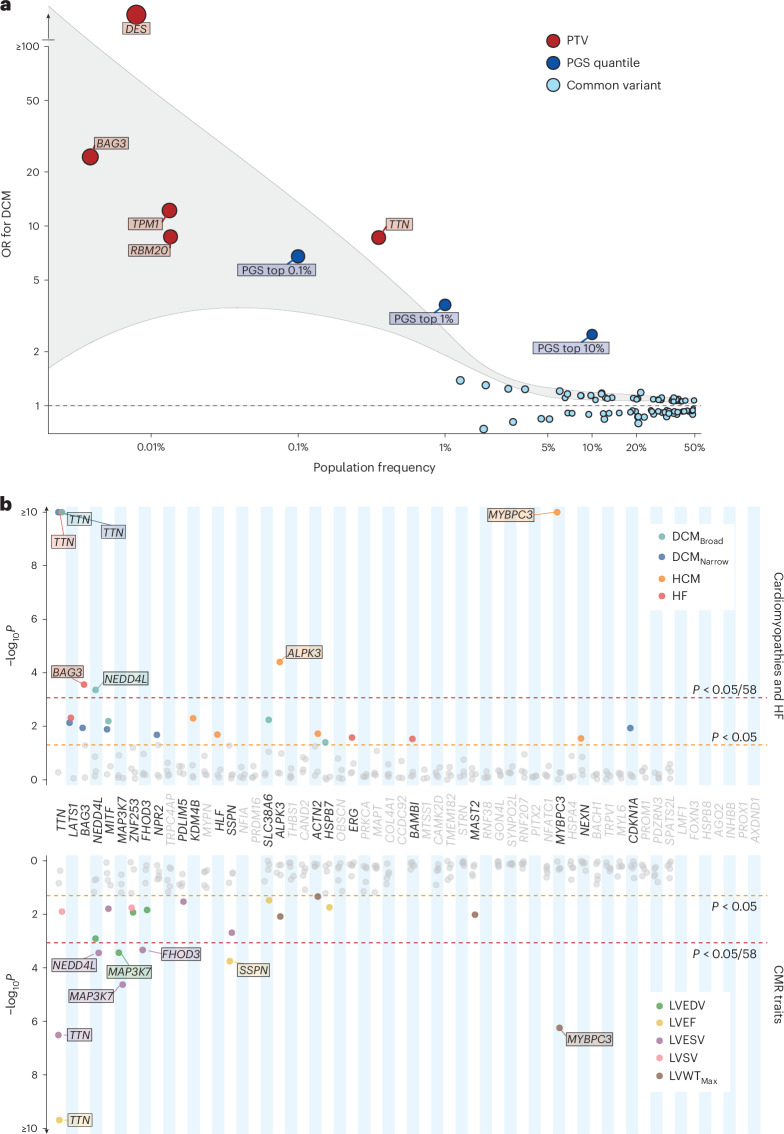
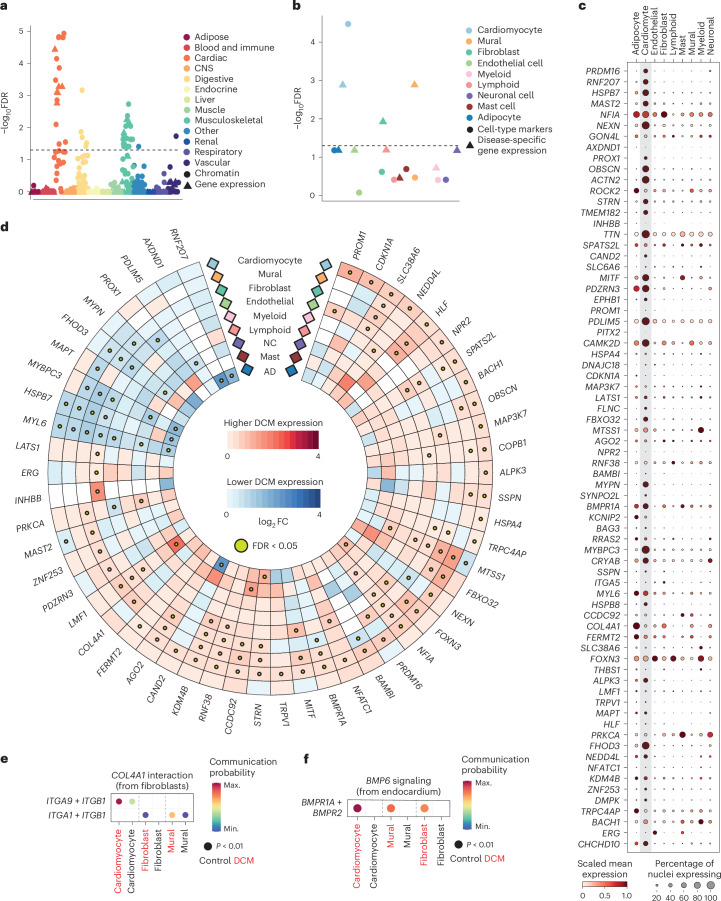
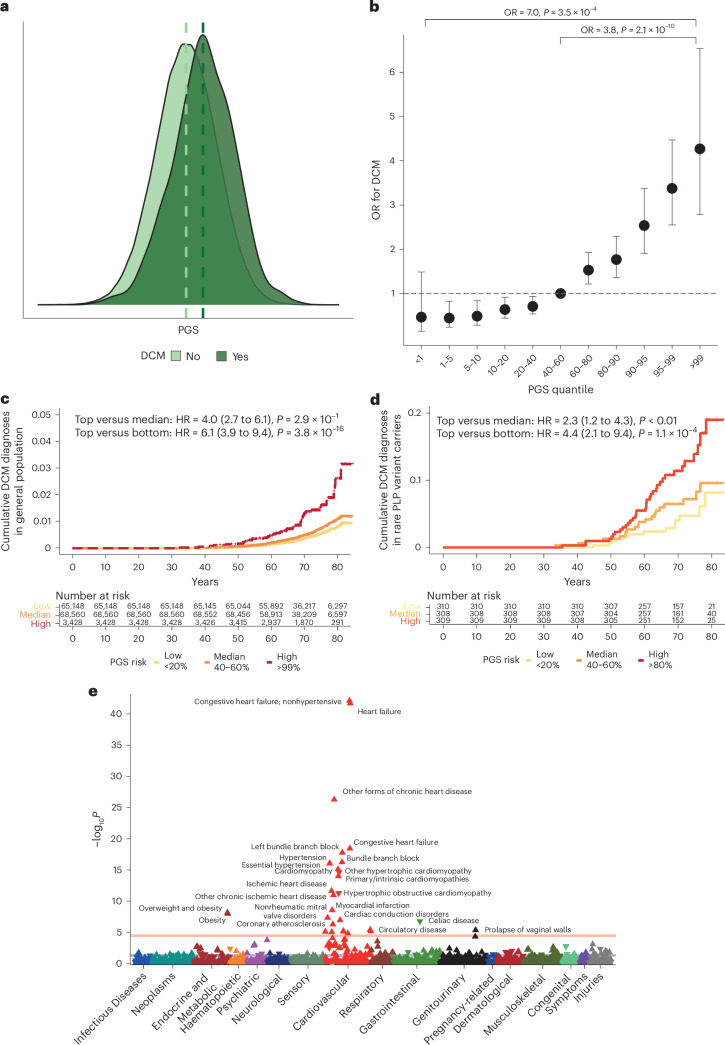
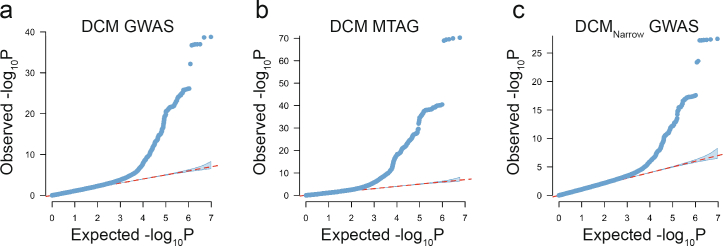
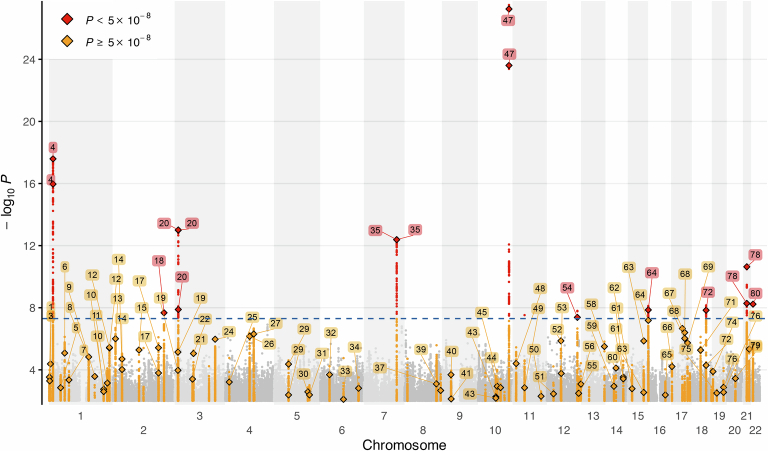
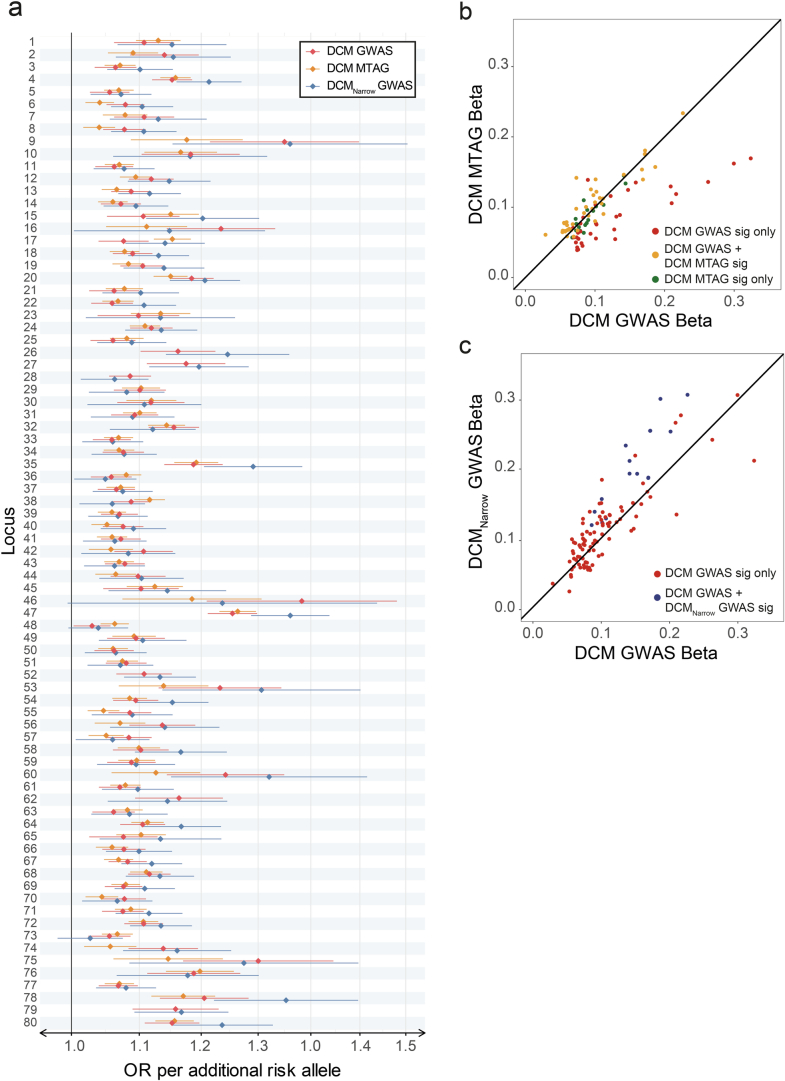
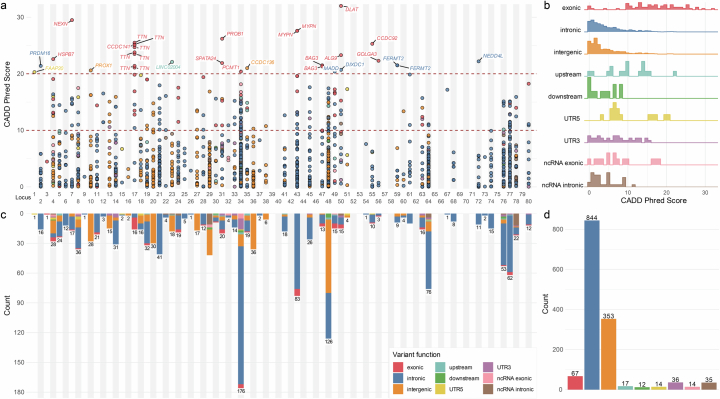
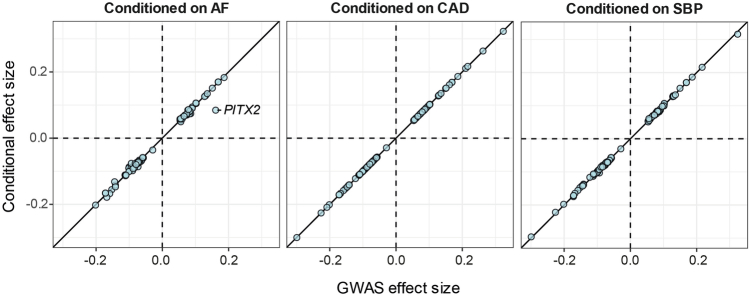
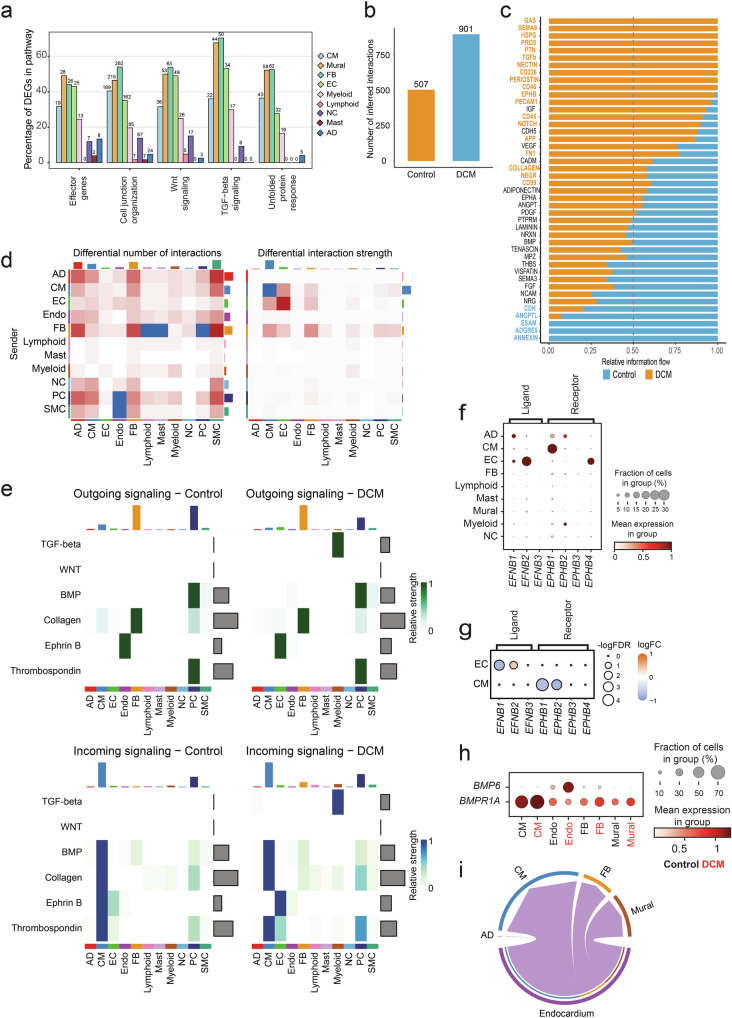
Dilated cardiomyopathy (DCM) is a leading cause of heart failure and cardiac transplantation. We report a genome-wide association study and multi-trait analysis of DCM (14,256 cases) and three left ventricular traits (36,203 UK Biobank participants). We identified 80 genomic risk loci and prioritized 62 putative effector genes, including several with rare variant DCM associations (MAP3K7, NEDD4L and SSPN). Using single-nucleus transcriptomics, we identify cellular states, biological pathways, and intracellular communications that drive pathogenesis. We demonstrate that polygenic scores predict DCM in the general population and modify penetrance in carriers of rare DCM variants. Our findings may inform the design of genetic testing strategies that incorporate polygenic background. They also provide insights into the molecular etiology of DCM that may facilitate the development of targeted therapeutics.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: S.L.Z. has acted as a consultant for Health Lumen. A.H. and R.T.L. have received funding from Pfizer Inc. R.T.L. has performed paid consultancy for Health Lumen and Fitfile Ltd. J.S.W. has acted as a consultant for MyoKardia, Pfizer, Foresite Labs and Health Lumen and received institutional support from Bristol Myers Squibb and Pfizer Inc. P.C. has received personal fees for consultancies, outside the present work, for Amicus, Pfizer Inc., Owkin and Bristol Myers Squibb. M.-P.D. declares holding equity in Dalcor Pharmaceuticals, unrelated to this work. The authors who are affiliated with deCODE genetics/Amgen Inc. and Regeneron Pharmaceuticals declare competing financial interests as employees. The other authors declare no competing interests.
Figures

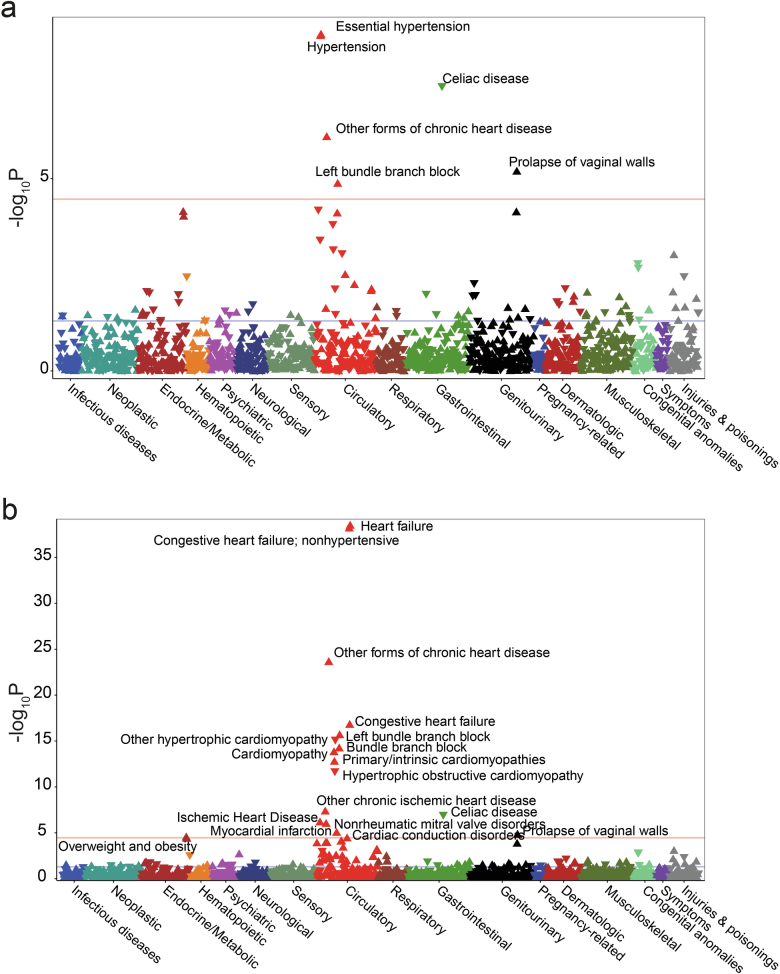
References
-
- Pinto, Y. M. et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J.37, 1850–1858 (2016). - DOI - PubMed
-
- Arbelo, E. et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J.44, 3503–3626 (2023). - PubMed
MeSH terms
Substances
Grants and funding
- FS/ICRF/24/26101/BHF_/British Heart Foundation/United Kingdom
- 207511/Z/17/Z/Wellcome Trust (Wellcome)
- SP/19/1/34461/BHF_/British Heart Foundation/United Kingdom
- MR/W023830/1/MRC_/Medical Research Council/United Kingdom
- FS/18/65/34186/British Heart Foundation (BHF)
- RG/19/6/34387/BHF_/British Heart Foundation/United Kingdom
- BBC/F/21/220106/BHF_/British Heart Foundation/United Kingdom
- MR/W026813/1/MRC_/Medical Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- RE/18/4/34215/British Heart Foundation (BHF)
- SP/17/11/32885/BHF_/British Heart Foundation/United Kingdom
- FS/ICRF/21/26019/BHF_/British Heart Foundation/United Kingdom
- MC_UP_1605/13/MRC_/Medical Research Council/United Kingdom
- FS/IPBSRF/22/27059/British Heart Foundation (BHF)
- MR/Y010175/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Miscellaneous
