

Genome-wide association study reveals mechanisms underlying dilated cardiomyopathy and myocardial resilience
- PMID: 39572784
- PMCID: PMC11631763
- DOI: 10.1038/s41588-024-01975-5
Genome-wide association study reveals mechanisms underlying dilated cardiomyopathy and myocardial resilience
Erratum in
-
Publisher Correction: Genome-wide association study reveals mechanisms underlying dilated cardiomyopathy and myocardial resilience.Nat Genet. 2024 Dec;56(12):2843. doi: 10.1038/s41588-024-02047-4. Nat Genet. 2024. PMID: 39633063 Free PMC article. No abstract available.
Abstract
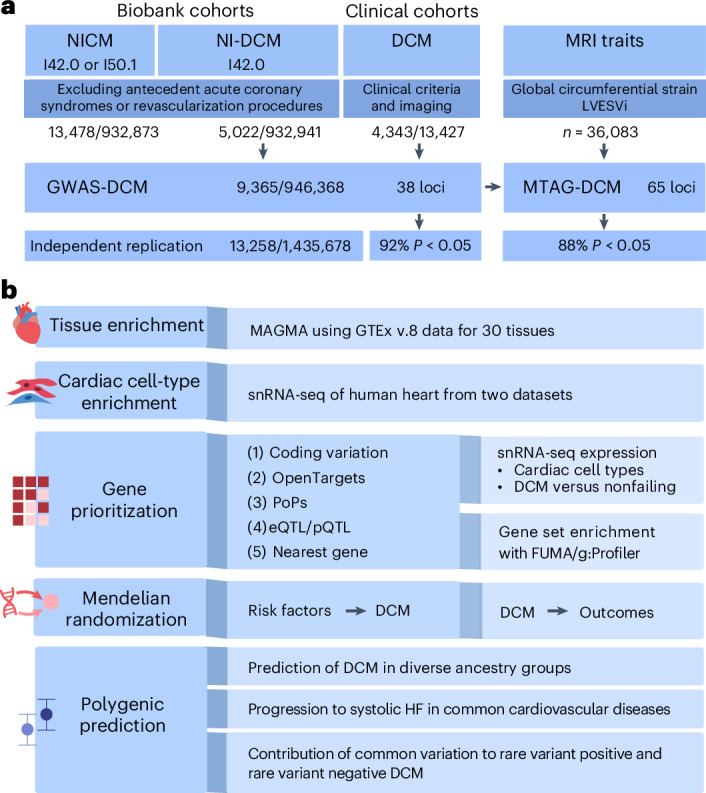
Dilated cardiomyopathy (DCM) is a heart muscle disease that represents an important cause of morbidity and mortality, yet causal mechanisms remain largely elusive. Here, we perform a large-scale genome-wide association study and multitrait analysis for DCM using 9,365 cases and 946,368 controls. We identify 70 genome-wide significant loci, which show broad replication in independent samples and map to 63 prioritized genes. Tissue, cell type and pathway enrichment analyses highlight the central role of the cardiomyocyte and contractile apparatus in DCM pathogenesis. Polygenic risk scores constructed from our genome-wide association study predict DCM across different ancestry groups, show differing contributions to DCM depending on rare pathogenic variant status and associate with systolic heart failure across various clinical settings. Mendelian randomization analyses reveal actionable potential causes of DCM, including higher bodyweight and higher systolic blood pressure. Our findings provide insights into the genetic architecture and mechanisms underlying DCM and myocardial function more broadly.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: P.T.E. has received sponsored research support from Bayer AG, IBM Health, Bristol Myers Squibb and Pfizer; he has consulted for Bayer AG, Novartis and MyoKardia. K.G.A. has received sponsored research support from Sarepta Therapeutics and Bayer AG, and reports a research collaboration with Novartis. Y.M.P. is involved in the development of therapies for DCM as an advisor to Forbion and Medical Director at ARMGO pharma and CMO at Phlox Therapeutics. P.C. reports personal fees for consultancies, outside the present work, for Amicus, OWKIN, Pfizer and SANOFI. J.S.W. has received research support from Bristol Myers Squibb, and has acted as a consultant for MyoKardia, Pfizer, Foresite Labs, Health Lumen and Tenaya Therapeutics. The remaining authors declare no competing interests.
Figures














References
MeSH terms
Grants and funding
- R01 HL139731/HL/NHLBI NIH HHS/United States
- I01-BX003362/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- R01 HL092577/HL/NHLBI NIH HHS/United States
- 18SFRN34110082/American Heart Association (American Heart Association, Inc.)
- 1RO1HL092577/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- 862032/American Heart Association (American Heart Association, Inc.)
- 521832260/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 1R01HL157635/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- RE/18/4/34215/British Heart Foundation (BHF)
- 1K08HL153937/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- I01 BX003362/BX/BLRD VA/United States
- 5R01HL139731/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- 21JTA/Sir Jules Thorn Charitable Trust
- SP/17/11/32885/British Heart Foundation (BHF)
- 03-007-2022-0035/Hartstichting (Dutch Heart Foundation)
- R01 HL157635/HL/NHLBI NIH HHS/United States
- 40-46800-98-018/ZonMw (Netherlands Organisation for Health Research and Development)
- K08 HL153937/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
