

Genetic heterogeneity in familial forms of genetic generalized epilepsy: from mono- to oligogenism
- PMID: 39574152
- PMCID: PMC11583555
- DOI: 10.1186/s40246-024-00659-9
Genetic heterogeneity in familial forms of genetic generalized epilepsy: from mono- to oligogenism
Abstract
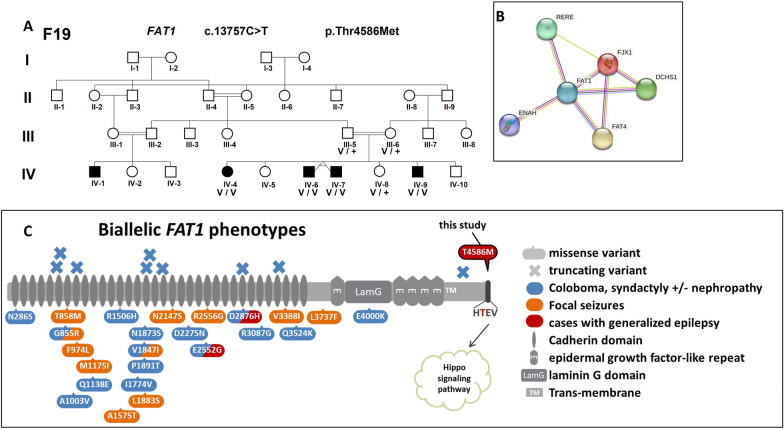
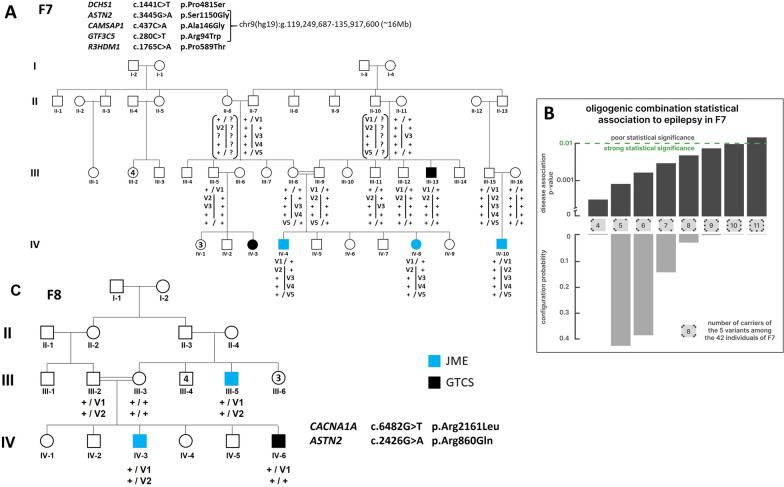
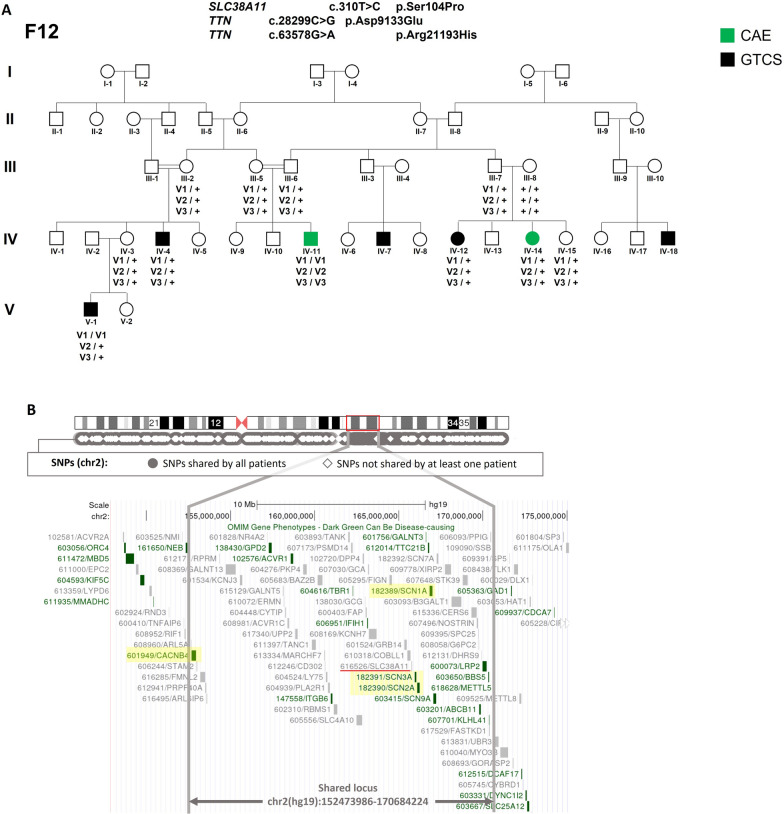
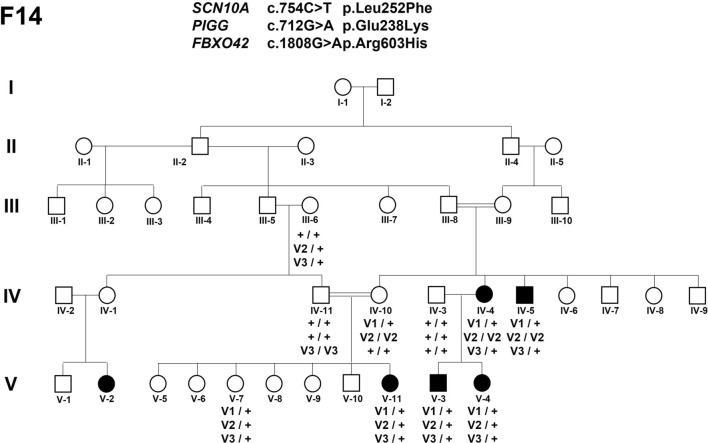
Genetic generalized epilepsy (GGE) including childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy (JME), and GGE with tonic-clonic seizures (TCS) (GGE-TCS), is genetically influenced with a two- to four- fold increased risk in the first-degree relatives of patients. Since large families with GGE are very rare, international studies have focused on sporadic GGE patients using whole exome sequencing, suggesting that GGE are highly genetically heterogeneous and rather involve rare or ultra-rare variants. Moreover, a polygenic mode of inheritance is suspected in most cases. We performed SNP microarrays and whole exome sequencing in 20 families from Sudan, focusing on those with at least four affected members. Standard genetic filters and Endeavour algorithm for functional prioritization of genes selected likely susceptibility variants in FAT1, DCHS1 or ASTN2 genes. FAT1 and DCHS1 are adhesion transmembrane proteins interacting during brain development, while ASTN2 is involved in dendrite development. Our approach on familial forms of GGE is complementary to large-scale collaborative consortia studies of sporadic cases. Our study reinforces the hypothesis that GGE is genetically heterogeneous, even in a relatively limited geographic area, and mainly oligogenic, as supported by the low familial penetrance of GGE and by the Bayesian algorithm that we developed in a large pedigree with JME. Since populations with founder effect and endogamy are appropriate to study autosomal recessive pathologies, they would be also adapted to decipher genetic components of complex diseases, using the reported bayesian model.
Keywords: ASTN2; DCHS1; FAT1; Bayesian model; GGE; JME; Oligogenic; Polygenic.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethical approval: This study was prospectively reviewed and approved by the national health research ethics committee, Federal ministry of health, Sudan (1–4–18). Consent to participate: Written informed consent was given by all participants. Consent for publication: Written informed consent was given by all participants. Competing interests: The authors declare no competing interests.
Figures
References
-
- Cossette P, Liu L, Brisebois K, Dong H, Lortie A, Vanasse M, Saint-Hilaire J-M, Carmant L, Verner A, Lu W-Y, Wang YT, Rouleau GA. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184–9. 10.1038/ng885. - PubMed
-
- Suzuki T, Delgado-Escueta AV, Aguan K, Alonso ME, Shi J, Hara Y, Nishida M, Numata T, Medina MT, Takeuchi T, Morita R, Bai D, Ganesh S, Sugimoto Y, Inazawa J, Bailey JN, Ochoa A, Jara-Prado A, Rasmussen A, Ramos-Peek J, Cordova S, Rubio-Donnadieu F, Inoue Y, Osawa M, Kaneko S, Oguni H, Mori Y, Yamakawa K. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36:842–9. 10.1038/ng1393. - PubMed
-
- Jara-Prado A, Martínez-Juárez IE, Ochoa A, González VM, Fernández-González-Aragón MDC, López-Ruiz M, Medina MT, Bailey JN, Delgado-Escueta AV, Alonso ME. Novel Myoclonin1/EFHC1 mutations in Mexican patients with juvenile myoclonic epilepsy. Seizure. 2012;21:550–4. 10.1016/j.seizure.2012.05.016. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
