

Performance of quantum chemistry methods for a benchmark set of spin-state energetics derived from experimental data of 17 transition metal complexes (SSE17)
- PMID: 39574537
- PMCID: PMC11577268
- DOI: 10.1039/d4sc05471g
Performance of quantum chemistry methods for a benchmark set of spin-state energetics derived from experimental data of 17 transition metal complexes (SSE17)
Abstract
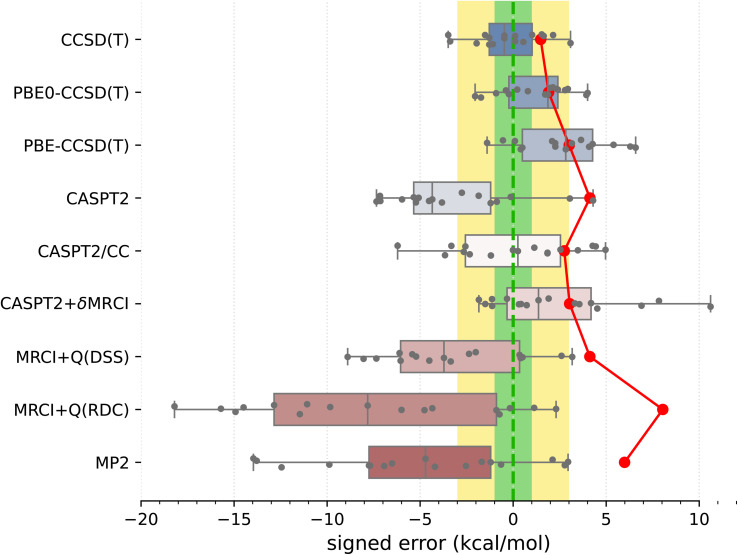
Accurate prediction of spin-state energetics for transition metal (TM) complexes is a compelling problem in applied quantum chemistry, with enormous implications for modeling catalytic reaction mechanisms and computational discovery of materials. Computed spin-state energetics are strongly method-dependent and credible reference data are scarce, making it difficult to conduct conclusive computational studies of open-shell TM systems. Here, we present a novel benchmark set of first-row TM spin-state energetics, which is derived from experimental data of 17 complexes containing FeII, FeIII, CoII, CoIII, MnII, and NiII with chemically diverse ligands. The estimates of adiabatic or vertical spin-state splittings, which are obtained from spin crossover enthalpies or energies of spin-forbidden absorption bands, suitably back-corrected for the vibrational and environmental effects, are employed as reference values for benchmarking density functional theory (DFT) and wave function methods. The results demonstrate a high accuracy of the coupled-cluster CCSD(T) method, which features the mean absolute error (MAE) of 1.5 kcal mol-1 and maximum error of -3.5 kcal mol-1, and outperforms all the tested multireference methods: CASPT2, MRCI+Q, CASPT2/CC and CASPT2+δMRCI. Switching from Hartree-Fock to Kohn-Sham orbitals is not found to consistently improve the CCSD(T) accuracy. The best performing DFT methods are double-hybrids (PWPB95-D3(BJ), B2PLYP-D3(BJ)) with the MAEs below 3 kcal mol-1 and maximum errors within 6 kcal mol-1, whereas the DFT methods so far recommended for spin states (e.g., B3LYP*-D3(BJ) and TPSSh-D3(BJ)) are found to perform much worse with the MAEs of 5-7 kcal mol-1 and maximum errors beyond 10 kcal mol-1. This work is the first such extensive benchmark study of quantum chemistry methods for TM spin-state energetics making use of experimental reference data. The results are relevant for the proper choice of methods to characterize TM systems in computational catalysis and (bio)inorganic chemistry, and may also stimulate new developments in quantum-chemical or machine learning approaches.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures



References
-
- Spin States in Bioinorganic and Inorganic Chemistry: Influence on Structure and Reactivity, ed. M. Swart and M. Costas, Wiley, 2015
LinkOut - more resources
Full Text Sources