

This is a preprint.
A PERTURBATION CELL ATLAS OF HUMAN INDUCED PLURIPOTENT STEM CELLS
- PMID: 39574586
- PMCID: PMC11580897
- DOI: 10.1101/2024.11.03.621734
A PERTURBATION CELL ATLAS OF HUMAN INDUCED PLURIPOTENT STEM CELLS
Abstract
Towards comprehensively investigating the genotype-phenotype relationships governing the human pluripotent stem cell state, we generated an expressed genome-scale CRISPRi Perturbation Cell Atlas in KOLF2.1J human induced pluripotent stem cells (hiPSCs) mapping transcriptional and fitness phenotypes associated with 11,739 targeted genes. Using the transcriptional phenotypes, we created a minimum distortion embedding map of the pluripotent state, demonstrating rich recapitulation of protein complexes, such as strong co-clustering of MRPL, BAF, SAGA, and Ragulator family members. Additionally, we uncovered transcriptional regulators that are uncoupled from cell fitness, discovering potential novel pluripotency (JOSD1, RNF7) and metabolic factors (ZBTB41). We validated these findings via phenotypic, protein-interaction, and metabolic tracing assays. Finally, we propose a contrastive human-cell engineering framework (CHEF), a machine learning architecture that learns from perturbation cell atlases to predict perturbation recipes that achieve desired transcriptional states. Taken together, our study presents a comprehensive resource for interrogating the regulatory networks governing pluripotency.
Conflict of interest statement
DECLARATION OF INTERESTS P.M. is a scientific co-founder of Shape Therapeutics, Boundless Biosciences, Navega Therapeutics, Pi Bio, and Engine Biosciences. The terms of these arrangements have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies. The Krogan Laboratory has received research support from Vir Biotechnology, F. Hoffmann-La Roche, and Rezo Therapeutics. Nevan Krogan has a financially compensated consulting agreement with Maze Therapeutics. Nevan Krogan is the President and is on the Board of Directors of Rezo Therapeutics, and he is a shareholder in Tenaya Therapeutics, Maze Therapeutics, Rezo Therapeutics, GEn1E Lifesciences, and Interline Therapeutics.
Figures
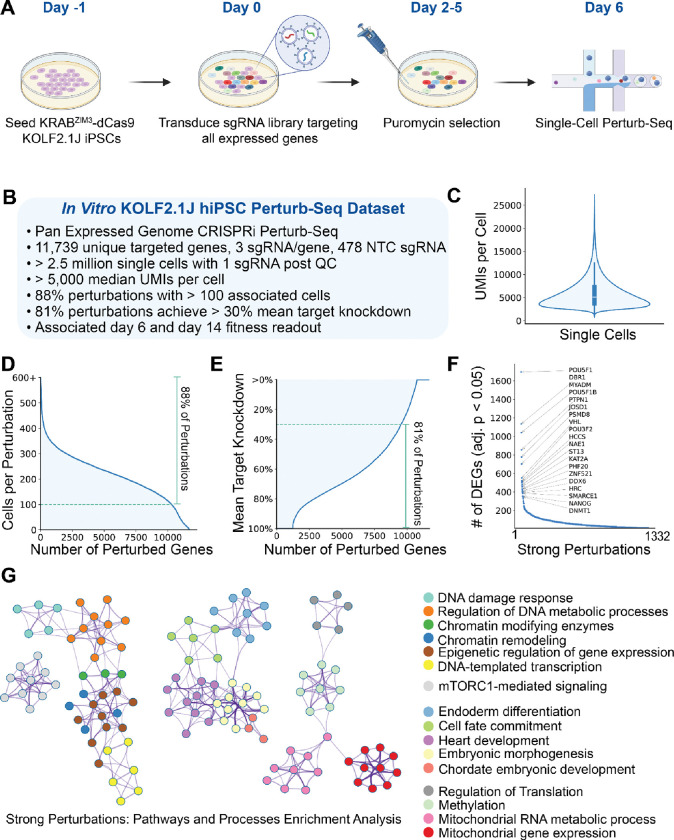
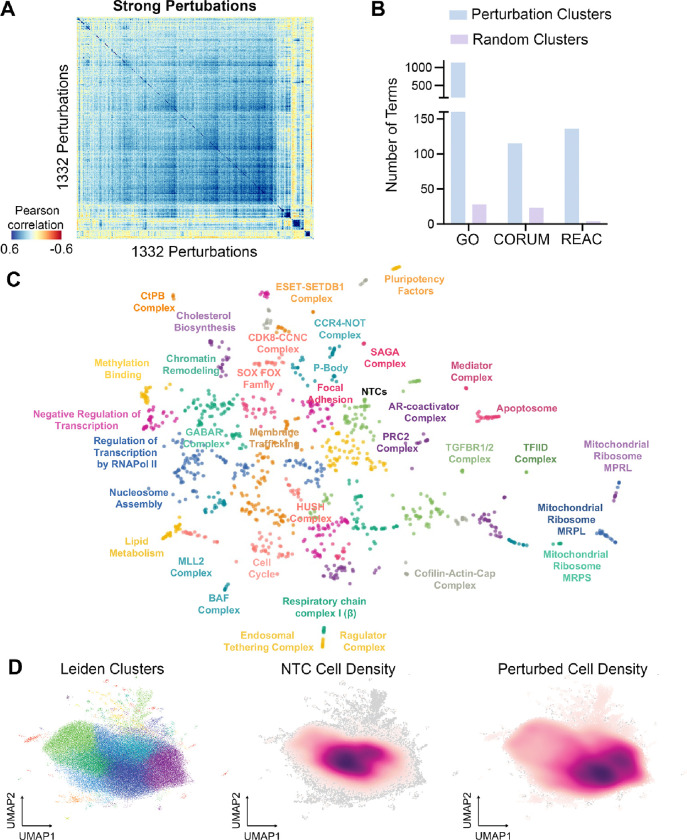


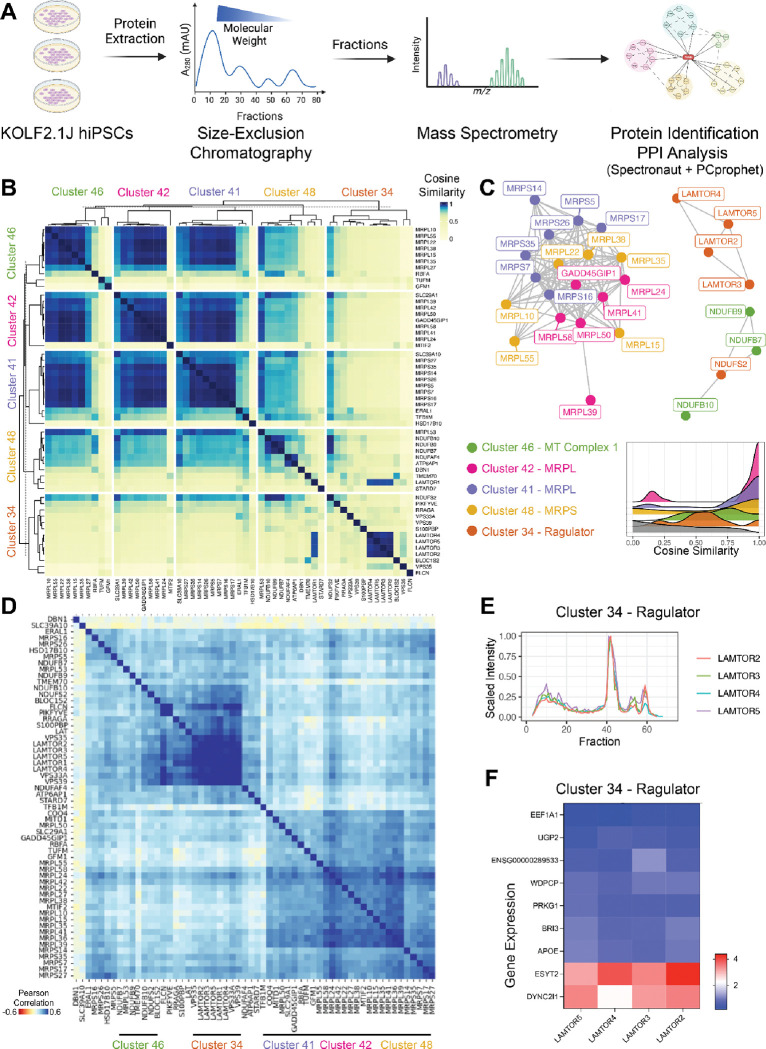

References
-
- Yamanaka S. (2020). Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 27, 523–531. - PubMed
-
- Park I.-H., Zhao R., West J.A., Yabuuchi A., Huo H., Ince T.A., Lerou P.H., Lensch M.W., and Daley G.Q. (2008). Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146. - PubMed
-
- Takahashi K., and Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources