

This is a preprint.
An mRNA Display Approach for Covalent Targeting of a Staphylococcus aureus Virulence Factor
- PMID: 39574702
- PMCID: PMC11581011
- DOI: 10.1101/2024.11.06.622387
An mRNA Display Approach for Covalent Targeting of a Staphylococcus aureus Virulence Factor
Update in
-
An mRNA Display Approach for Covalent Targeting of a Staphylococcus aureus Virulence Factor.J Am Chem Soc. 2025 Mar 12;147(10):8312-8325. doi: 10.1021/jacs.4c15713. Epub 2025 Feb 27. J Am Chem Soc. 2025. PMID: 40013487
Abstract
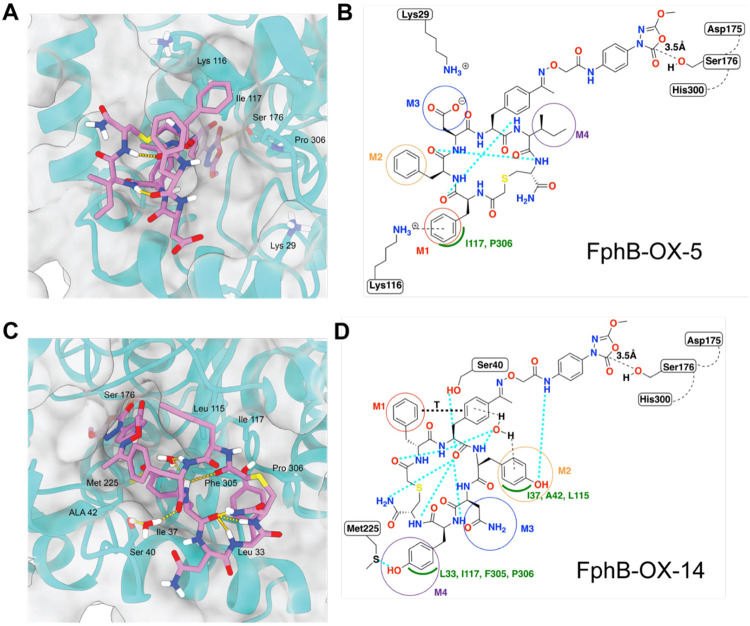
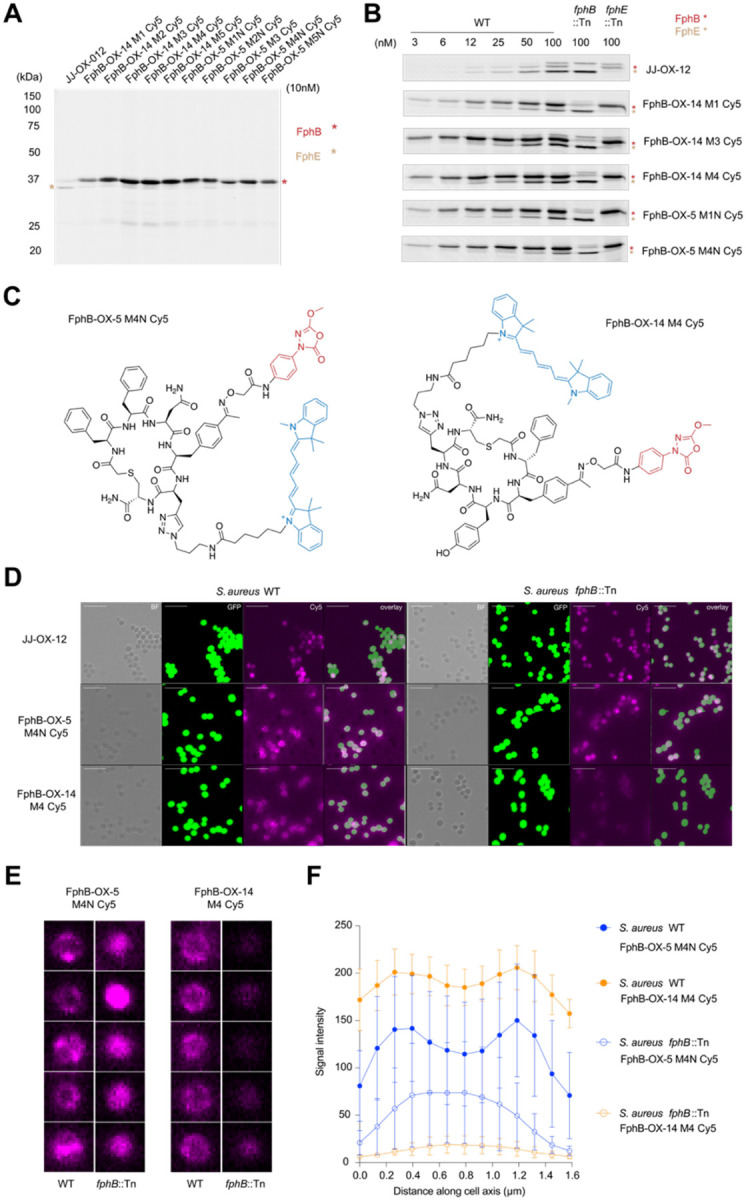
Staphylococcus aureus (S. aureus) is an opportunistic human pathogen that causes over one million deaths around the world each year. We recently identified a family of serine hydrolases termed fluorophosphonate binding hydrolases (Fphs) that play important roles in lipid metabolism and colonization of a host. Because many of these enzymes are only expressed in Staphylococcus bacteria, they are valuable targets for diagnostics and therapeutics. Here we developed and screened highly diverse cyclic peptide libraries using mRNA display with a genetically encoded oxadiazolone (Ox) electrophile that was previously shown to potently and covalently inhibit multiple Fph enzymes. By performing multiple rounds of counter selections with WT and catalytic dead FphB, we were able to tune the selectivity of the resulting selected cyclic peptides containing the Ox residue towards the desired target. From our mRNA display hits, we developed potent and selective fluorescent probes that label the active site of FphB at single digit nanomolar concentrations in live S. aureus bacteria. Taken together, this work demonstrates the potential of using direct genetically encoded electrophiles for mRNA display of covalent binding ligands and identifies potent new probes for FphB that have the potential to be used for diagnostic and therapeutic applications.
Figures
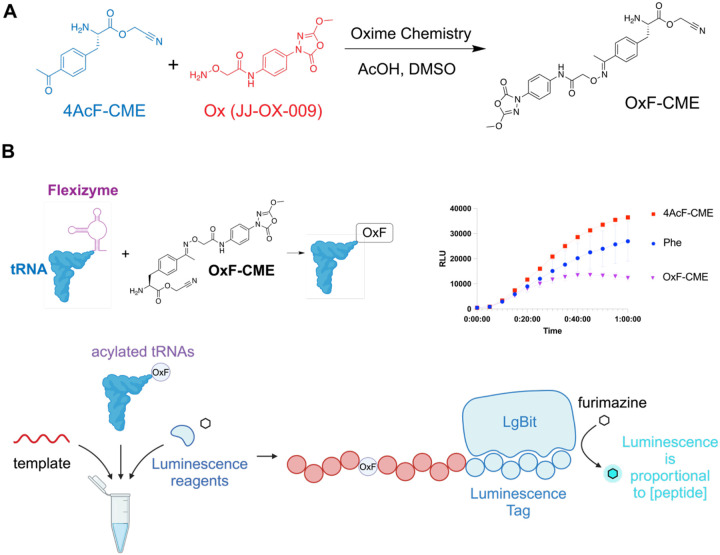
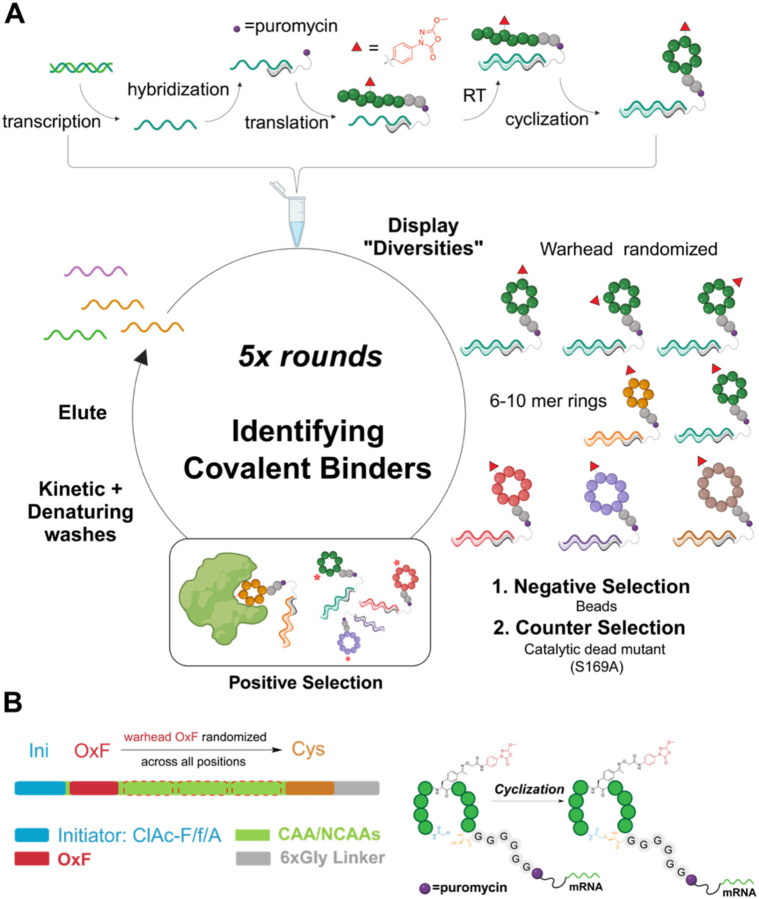
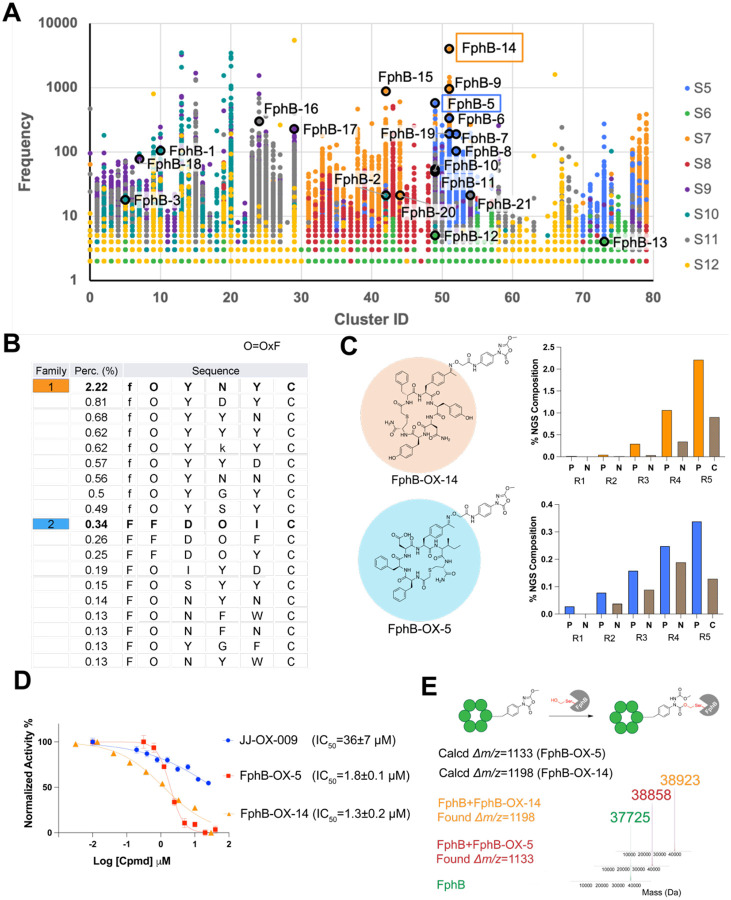
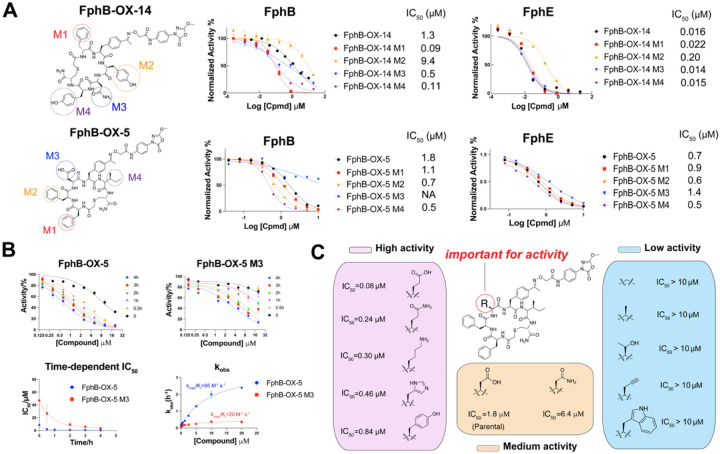
References
-
- Schwartz P. A.; Kuzmic P.; Solowiej J.; Bergqvist S.; Bolanos B.; Almaden C.; Nagata A.; Ryan K.; Feng J.; Dalvie D.; Kath J. C.; Xu M.; Wani R.; Murray B. W. Covalent EGFR Inhibitor Analysis Reveals Importance of Reversible Interactions to Potency and Mechanisms of Drug Resistance. Proceedings of the National Academy of Sciences 2014, 111 (1), 173–178. 10.1073/pnas.1313733111. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources