

This is a preprint.
Addressing the mean-variance relationship in spatially resolved transcriptomics data with spoon
- PMID: 39574747
- PMCID: PMC11580860
- DOI: 10.1101/2024.11.04.621867
Addressing the mean-variance relationship in spatially resolved transcriptomics data with spoon
Update in
-
Addressing the mean-variance relationship in spatially resolved transcriptomics data with spoon.Biostatistics. 2024 Dec 31;26(1):kxaf012. doi: 10.1093/biostatistics/kxaf012. Biostatistics. 2024. PMID: 40515599 Free PMC article.
Abstract
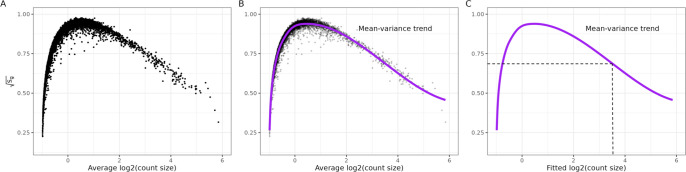
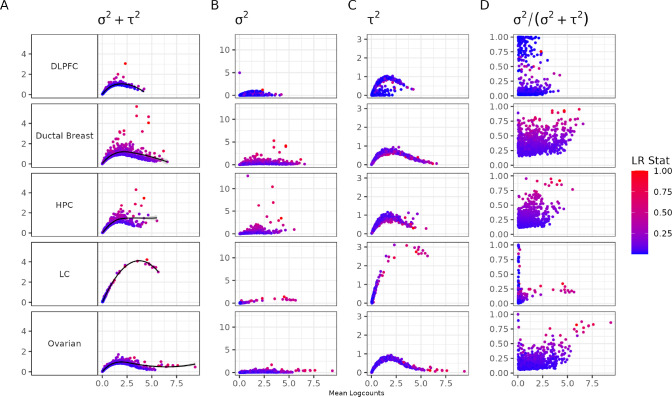
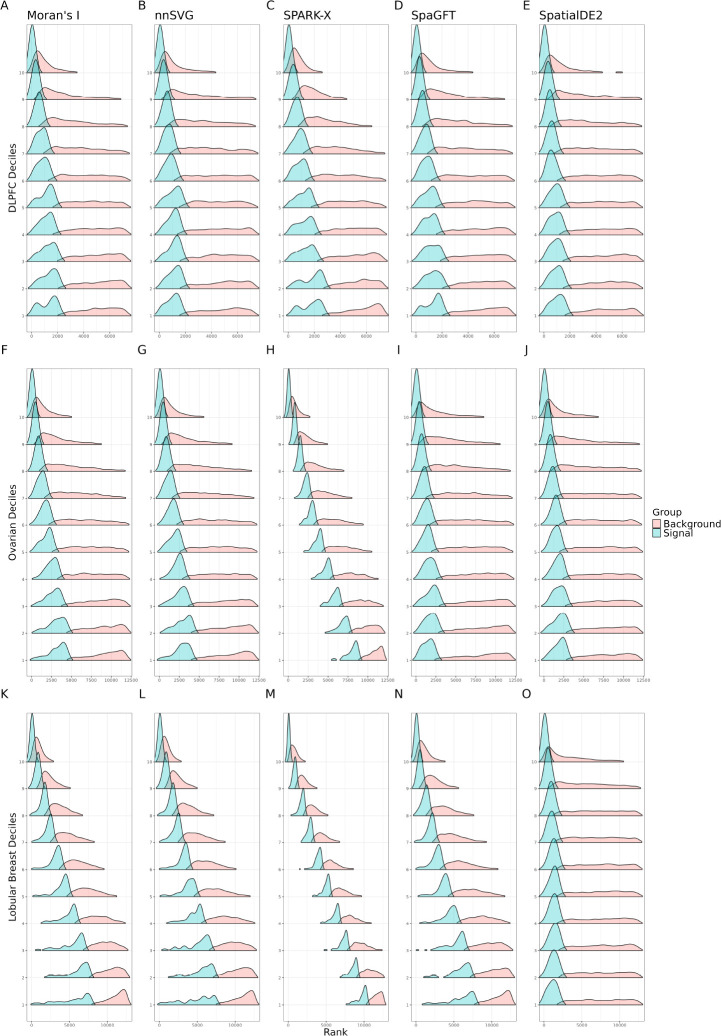
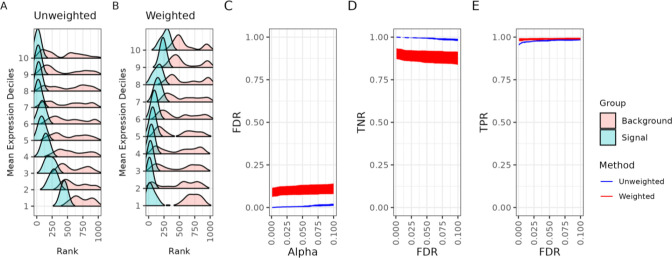
An important task in the analysis of spatially resolved transcriptomics data is to identify spatially variable genes (SVGs), or genes that vary in a 2D space. Current approaches rank SVGs based on either p-values or an effect size, such as the proportion of spatial variance. However, previous work in the analysis of RNA-sequencing identified a technical bias, referred to as the "mean-variance relationship", where highly expressed genes are more likely to have a higher variance. Here, we demonstrate the mean-variance relationship in spatial transcriptomics data. Furthermore, we propose spoon, a statistical framework using Empirical Bayes techniques to remove this bias, leading to more accurate prioritization of SVGs. We demonstrate the performance of spoon in both simulated and real spatial transcriptomics data. A software implementation of our method is available at https://bioconductor.org/packages/spoon.
Keywords: Gaussian process regression; empirical Bayes; mean-variance bias; spatial transcriptomics; spatially variable gene.
Conflict of interest statement
Conflict of Interest: None declared.
Figures
References
-
- Marx V.. Method of the Year: spatially resolved transcriptomics. Nature Methods, 18(1):9–14, Jan. 2021. ISSN 1548–7091, 1548–7105. doi: 10.1038/s41592-020-01033-y. URL https://www.nature.com/articles/s41592-020-01033-y. - DOI - PubMed
-
- Deshpande A., Loth M., Sidiropoulos D. N., Zhang S., Yuan L., Bell A. T., Zhu Q., Ho W. J., Santa-Maria C., Gilkes D. M., Williams S. R., Uytingco C. R., Chew J., Hartnett A., Bent Z. W., Favorov A. V., Popel A. S., Yarchoan M., Kiemen A., Wu P.-H., Fujikura K., Wirtz D., Wood L. D., Zheng L., Jaffee E. M., Anders R. A., Danilova L., Stein-O’Brien G., Kagohara L. T., and Fertig E. J.. Uncovering the spatial landscape of molecular interactions within the tumor microenvironment through latent spaces. Cell Systems, 14(4):285–301.e4, Apr. 2023. ISSN 24054712. doi: 10.1016/j.cels.2023.03.004. URL https://linkinghub.elsevier.com/retrieve/pii/S2405471223000807. - DOI - PMC - PubMed
-
- Rao A., Barkley D., França G. S., and Yanai I.. Exploring tissue architecture using spatial transcriptomics. Nature, 596(7871):211–220, Aug. 2021. ISSN 0028–0836, 1476–4687. doi: 10.1038/s41586-021-03634-9. URL https://www.nature.com/articles/s41586-021-03634-9. - DOI - PMC - PubMed
-
- Garcia-Alonso L., Lorenzi V., Mazzeo C. I., Alves-Lopes J. P., Roberts K., Sancho-Serra C., Engelbert J., Marečková M., Gruhn W. H., Botting R. A., Li T., Crespo B., Van Dongen S., Kiselev V. Y., Prigmore E., Herbert M., Moffett A., Chédotal A., Bayraktar O. A., Surani A., Haniffa M., and Vento-Tormo R.. Single-cell roadmap of human gonadal development. Nature, 607(7919):540–547, July 2022. ISSN 0028–0836, 1476–4687. doi: 10.1038/s41586-022-04918-4. URL https://www.nature.com/articles/s41586-022-04918-4. - DOI - PMC - PubMed
-
- Chen K. S., Noureldein M. H., Rigan D. M., Hayes J. M., Savelieff M. G., and Feldman E. L.. Regional interneuron transcriptional changes reveal pathologic markers of disease progression in a mouse model of Alzheimer’s disease, Nov. 2023. URL 10.1101/2023.11.01.565165v1. - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources